转录因子Nrf2在缺氧性肺血管重构中的作用机制
2021-07-09陈依林李光才
陈依林 李光才
转录因子是抗氧化反应的重要的上游调节剂,它可以维持细胞氧化还原稳态并减少严重的氧化损伤[1]。在最近的20年中,核因子-E2相关因子2(Nuclear factor-E2-related factor 2,Nrf2)的发现与氧化应激在许多关键疾病的发病机制中作用的证据不断增加[2]。Nrf2稳态受细胞质抑制剂Kelch样ECH相关蛋白1的调节,以响应内部清洁蛋白需求或氧化剂,异源生物、致癌物、抗氧化剂和化学预防剂的外源刺激,不仅有助于维持细胞氧化还原平衡,而且还有助于调节细胞周期和死亡、免疫力、新陈代谢、选择性蛋白质降解、发育和致癌作用[3]。小肠缺血/再灌注(intestinal ischemia/reperfusion,IIR)术中并发症可引起继发性血管疾病和许多相关的生理状况,主要与缺血组织和炎性介质释放的细胞毒性物质有关[4]。因此,IIR可能导致败血症,全身性炎性反应综合征和多器官功能障碍综合征[5]。已知Nrf2可以激活各种抗氧化反应元件依赖性基因响应氧化应激[6]。Nrf2也参与调节视网膜和其他组织床的缺血性血管新生[7]。Nrf2被认为在缺氧时会增加炎症和损伤,从而在与氧化应激相关的肺损伤中起保护作用[8]。Sirt1是依赖烟酰胺腺嘌呤二核苷酸的脱乙酰酶蛋白酶,被认为通过脱酰作用以及与几个转录因子和信号分子的相互作用而参与了许多调控过程[9]。上调Sirt1的水平会增加Nrf2的核易位,进而抑制丝裂原激活的蛋白激酶磷酸化,并提供针对氧化应激引起的细胞损伤的保护作用[10]。本研究探讨Nrf2/Sirt1通路在调节氧-葡萄糖剥夺/复氧(Oxygen-glucose deprivation/reoxygenation,OGD/R)后肺损伤后inNrf2缺乏的Nrf2敲低人肺微血管内皮细胞(Pulmonary microvascular endothelial cells,PMVEC)的血管重建和急性肺损伤的调节。
1 资料与方法
1.1 一般资料
1.1.1 实验大鼠:在实验中使用C57BL/6J小鼠(周龄10周左右)和Nrf2基因敲除(Nrf2-/-)的小鼠,所有小鼠具有相同的遗传背景。使用日本MEXT的Project进行体内实验。以12 h的明/暗周期将小鼠保持在受控温度(21±2)℃和湿度(60±5)%下。小鼠可随意获取标准食物。
1.1.2 小鼠肺损伤模型构建:用戊巴比妥钠(50 mg/kg,腹腔注射)麻醉,并在手术过程中自发呼吸。通过中线切口打开麻醉小鼠的腹壁。肠系膜上动脉暴露并被动脉瘤夹夹住。到肠的血流中断90 min,释放以重新建立血流。假手术组进行相同的手术,除了夹住肠系膜上动脉。手术后腹膜内注射0.9%氯化钠溶液(1.0 ml)使所有小鼠复苏。再灌注6 h后处死动物并收获组织。将组织在液氮中速冻并保存在-80℃直至分析。
1.1.3 实验分组:将实验小鼠随机分配到以下四组(每组n=10):WT小鼠对照组(野生型小鼠不做手术处理);WT小鼠损伤组(该组小鼠进行肠缺血/再灌注手术处理,进行肺损伤模型构建);Nrf2-/-小鼠损伤组(Nrf2-/-小鼠进行肠缺血/再灌注手术处理);Nrf2 siRNA转染组(Nrf2抑制表达);Sirt1过表达组(用Sirt1慢病毒载体转染PMVEC)。
1.1.4 医学伦理学问题:所有实验程序均经动物保护和使用委员会批准的方案下进行,并根据实验动物护理和使用指南进行实验。
1.2 实验方法
1.2.1 CCK-8测定细胞增殖:细胞计数试剂盒8(Sigma,日本)用于检测NSCLC细胞的增殖。将细胞以每孔5 000个细胞的密度接种在96孔板中,并在5%CO2中于37℃的培养箱中培养2 h以粘附细胞。向每个孔中添加10 μl细胞增殖试剂CCK-8,混合,在培养箱中培养2 h。双波长酶标仪用于测量450~490 nm 的检测波长和600~650 nm的参考波长(贝克曼库尔特,美国)。每个实验都设置了3个平行重复。
1.2.2 细胞培养及转染:人肺微血管内皮细胞的氧葡萄糖剥夺和复氧人PMVEC从科学细胞研究实验室(目录号:3 000)购买。为了进行OGD/R,将细胞在无葡萄糖的DMEM中培养。将它们在补充有0.5 mmol/L CaCl2和1 mmol/L MgCl2的PBS中洗涤,并置于厌氧室(5%CO2、95%N2; Memmert; Schwabach,德国)中以诱导OGD。8 h后,将培养基替换为ECM,并使平板在常氧室(37℃,5%CO2)中生长,进行24 h的复氧。Sirt1载体对PMVEC的瞬时转染,如先前所述,用Sirt1慢病毒载体转染PMVEC。将PMVECs以每孔2×105的密度铺在6孔板中,感染感染复数(MOI)为20的慢病毒载体。通过在10 ml的分选缓冲液(含2%FBS,1 mmol/L EDTA的D-PBS)和50 U/ml DNAse中上下吸打,将肺组织解离。样品在摇动下于37℃孵育10 min,通过100 μm,70 μm和40 μm细胞过滤器转移到50 ml管中。将过滤后的悬浮液转移至15 ml试管中,并在4℃下以550×g的转速离心5 min以沉淀细胞。除去上清液,并将细胞重悬于10 ml分选缓冲液中,并通过在37℃下振荡孵育1 h使其恢复。
1.2.3 组织学检查:在4 mm苏木精和曙红(HE)染色的石蜡包埋切片中确定缺血-再灌注损伤。根据肺泡充血、出血、水肿和炎性细胞在空域或血管壁浸润的综合评估,使用五点量表对急性肺损伤进行评分。盲目使用0-4(最低至最高严重性)评分系统。肺纤维化是通过使用Image Pro-Plus 6.0软件对Masson的Trichrome阳性染色区域进行定量分析来测量的。图像是用连接与数码相机(Optronics DEI-470;加利福尼亚州戈莱塔)捕获的。使用评分系统评估肺损伤程度。根据该评分系统,将肺泡和间充质的水肿,肺泡内炎性细胞浸润,肺泡出血和肺不张的等级分为0~4级。等级:0级,正常,<所占空间的15%被组织和> 85%的肺泡间隙占据;1级,15%~25%的空间被组织占据,而76%~85%的空间被肺泡占据;2级,组织占26%~50%,肺泡间隙占51%~75%;3级,组织占51%~75%,肺泡间隙占26%~50%;4级,组织占76%~100%,肺泡间隙占0~25%。
1.2.4 蛋白质印迹分析:通过先前描述的方法从PMVEC制备核提取物。通过刮擦在含有磷酸酶抑制剂的冰冷的PBS中收集细胞,并通过离心沉淀在1 000×g下5 min。将沉淀重悬于500 ml 1×低渗缓冲液中,在冰上孵育15 min,添加去污剂(25 ml)并高速涡旋30 s。将悬浮液在4℃离心(14 000×g,20 min);将核沉淀重悬于裂解缓冲液中(50 ml),并在冰上孵育15 min。将该悬浮液进一步离心(14 000×g)10 min,将等分的上清液(核提取物)储存在-80℃进行进一步分析。通过BCA蛋白质测定法定量核提取物中的蛋白质含量。蛋白质印迹分析蛋白质印迹通过标准规程进行。通过SDS-PAGE分离总细胞裂解液或肺组织中的蛋白质(30 mg),并转移到硝酸纤维素膜上。通过化学发光(ECL;Amersham)观察条带,暴露于X射线胶片并使用ImageJ进行光密度测定定量。
1.2.5 肺胶原定量:切断腹主动脉后,用无菌0.9%氯化钠溶液(15 ml)灌注肺血管,在肺门切开左肺,匀浆,在65%三氯乙酸中孵育(Fisher Scientific,宾夕法尼亚州匹兹堡)在冰上。离心并吸出上清液后,将组织沉淀重悬于12 N HCl中,并在110℃下烘烤过夜。将变性的组织在ddH2O中重新配制,将等分试样(200 ml)与在10%异丙醇和0.5 mmol/L乙酸钠中的1.4%氯胺T于室温孵育20 min。加入1 mmol/L 4-(二甲基氨基)-苯甲醛(500 ml; Sigma-Aldrich),并将该悬浮液在65℃下温育15 min。对于每个样品,一式三份地测量540 nm处的吸光度。 从氯胺反应开始,同时使用0~500 mg/ml反式4-羟基-L-脯氨酸(Sigma-Aldrich)同时生成标准曲线。
1.2.6 RNA提取和实时PCR:使用RNeasy minikit(QIAGEN,德国汉堡)提取总RNA。对于实时PCR,将RNA(2 mg)反转录成互补DNA(40 ng)的模板。放大循环反应(40个循环)如下进行:在95℃下5 s和在60℃下30 s。根据比较阈值循环(2-DDCT)标准化靶基因的相对定量值。见表1。

表1 RT-PCR引物序列

2 结果
2.1 肺损伤后Nrf2的表达 通过免疫荧光染色确定肺伤后Nrf2的表达,WT小鼠损伤组较WT小鼠对照组Nrf2的荧光染色增加(P<0.05)。说明在肺损伤后Nrf2在肺组织的细胞核中大量积聚。见表2。

表2 免疫荧光染色确定Nrf2的表达
2.2 蛋白质印迹分析 通过蛋白质印迹分析实验小鼠的HIF-1α、VEGF和CD31的蛋白表达,WT小鼠损伤组较Nrf2-/-小鼠损伤组HIF-1α、VEGF和CD31的蛋白表达升高(P<0.05)。表明Nrf2有利于改善血管生成。见表3。
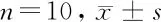
表3 蛋白印迹分析
2.3 Nrf2对肺损伤后的调节 WT小鼠损伤组较Nrf2-/-小鼠损伤组白细胞浸润量降低(P<0.05),Nrf2-/-小鼠损伤组较WT小鼠损伤组肺损伤评分升高(P<0.05),WT小鼠损伤组较Nrf2-/-小鼠损伤组胶原蛋白沉淀降低(P<0.05)。见表4。
表4 实验小鼠的肺损伤指标检
2.4 细胞增殖检测 第12小时 Sirt1过表达组较Nrf2 siRNA转染组细胞增殖升高(P<0.05),第24小时Nrf2 siRNA转染组较 Sirt1过表达组细胞增殖降低(P<0.05)。见表5。

表5 细胞增殖检测
2.5 RT-PCR分析 通过PCR实时分析细胞的CD31、NOX4和Nrf2的mRNA表达,Sirt1过表达组较Nrf2 siRNA转染组CD31、NOX4和Nrf2的mRNA表达升高(P<0.05)。表明 Sirt1的过表达可诱导Nrf2上调,促进NOX4介导的基因调控促进人类PMVEC中的血管生成。见表6,图1。
表6 RT-PCR实时分析CD31、NOX4和Nrf2的mRNA表达
3 讨论
Nrf2是抗氧化反应元件的基因的关键转录因子,参与多种宿主防御功能,包括氧化还原平衡,细胞周期,免疫力,线粒体生物发生,能量代谢和致癌作用[11]。呼吸道中的Nrf2尤其重要,因为呼吸系统会不断与环境压力相接[12]。由于Nrf2被确定为模型急性肺损伤的易感基因,因此在人类疾病的实验模型中使用易补充呼吸治疗(例如高氧血症,机械通气)的Nrf2突变小鼠已证明了Nrf2在气道中的保护能力[13]。Nrf2已参与调节多种疾病的缺血性血管新生,例如心血管疾病和缺血性视网膜病[14]。Nrf2也被认为可以保护许多器官免受IIR伤害,包括肺。此外,在几项研究中,发现Sirt1的上调可激活Nrf2[15]。我们研究了Nrf2是否参与了IIR在鼠模型中急性肺损伤的血管形成和调节,发现Sirt1过表达刺激的Nrf2可能在缺血后的血管新生中起重要作用IIR通过NOX4介导的基因调控。由Sirt1过表达刺激的Nrf2上调和易位向组织病理学变化的缓解并增强了内皮细胞标志物CD31的表达。Nrf2-抗氧化反应元件信号通路是抵抗氧化或亲电子应激的主要细胞防御手段[16]。它通过调节诱导型和组成型基因的表达与许多抗氧化反应元件依赖基因相互作用[17]。深入了解抗氧化反应元件诱导型/组成型基因的调控可能会导致以控制所需的质量,例如缺血组织的血运重建[18]。我们发现在小鼠中Nrf2敲低抑制了IIR后的CD31,HIF-1α和VEGF表达,而Sirt1对Nrf2的上调增强了野生型IIR后的CD31,HIF-1α和VEGF表达。HIF-1α参与VEGF的转录,从而刺激缺氧或局部缺血后的新生血管形成[19]。使用视网膜病变的大鼠模型研究了新生血管形成,发现VEGF激活的NOX4导致了VEGF活化的VEGFR2和NOX4介导内皮细胞增殖,并提出Sirt1可以发挥血管生成的作用,发现Sirt1通过FOXO3信号通路对IIR诱导的急性肺损伤具有保护作用[20]。为了研究与Nrf2/Sirt1相关的血管生成,本研究评估了OGD/R后人PMVEC中Nrf2和Sirt1的调控。发现Sirt1过表达诱导Nrf2上调并易位到核内。Nrf2的核积累与增强的Sirt1介导的CD31、NOX4和Nrf2表达有关。此外,Sirt1在细胞质和核区室之间穿梭,并调节对细胞的生长,细胞存活和迁移相关的基因表达,以响应生理和病理性细胞应激。因此,Nrf2/Sirt1我们在鼠模型中观察到的调节血管生成在人肺内皮细胞中复制。
总之,我们发现Sirt1的过表达促进了Nrf2向核的转运,并增加了鼠和人细胞模型中NOX4、HIF-1α和VEGF的表达。这导致鼠模型中的缺血性血管生成和增加了人类细胞中的细胞活力。当受Sirt1刺激时,Nrf2可能通过NOX4介导的基因调控在血管生成中发挥重要作用。
