原料药中基因毒性杂质分类清除及控制策略
2021-07-07关元宙梁毅
关元宙,梁毅
(中国药科大学国际医药商学院,南京 211198)
原料药的生产、运输及储存过程中可能引入杂质,这不仅影响药物纯度,还可能引起药物不良反应,其中基因毒性杂质能以较低水平浓度导致体内DNA受损突变,进而引发癌症,对患者用药安全危害大。经过十几年的发展,药品监管部门及医药行业对药品中基因毒性杂质的性质、清除及控制均有了新的认知,各国药品监管部门通报的基因毒性杂质相关药品召回事件也引起了工业界及大众对这类杂质的关注。基于现行ICH 指导原则与行业实施现状,本文综合基因毒性杂质特点、分类控制原则、风险评估方法、原料药生产工艺及质量标准等方面阐述这类杂质的控制策略。
1 原料药中的基因毒性杂质
原料药合成过程中使用到多种起始物料、试剂及溶剂,反应过程中还存在副反应及中间体残留,原料药分子在运输储存过程中还受时间、光照、温度、pH 值、水、辅料及直接接触包材的影响而发生降解,以上情况均有可能向原料药体系引入潜在基因毒性杂质,见图1。在原料药杂质谱建立过程中,通常需要对合成路线中原料、中间体、副产物及后续降解产物进行基因毒性分析。常用鉴定杂质具有基因毒性的方法:首先筛选杂质结构,确认存在已知基因毒性杂质相似致突变官能团或经计算机结构毒理学评估程序判定具有基因毒性结构的,再经基因毒性检测(如Ames 测试)得到阳性结果,有时辅以体内检测最终确认杂质基因毒性[1]。
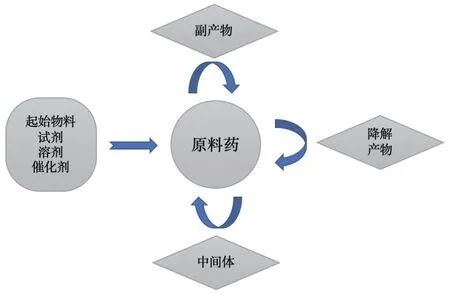
图1 原料药中基因毒性杂质引入途径Fig.1 Approaches in genotoxic impurities introducing to drug substances
2 潜在基因毒性化合物相关反应
常见的基因毒性物质具有相似的某几类官能团,这些官能团常作为警示结构预估杂质是否具有基因毒性。具有警示结构的物质常作为反应物质或反应介质参与原料药的合成,本文归纳了常见警示结构化合物在原料药合成过程中为体系引入基因毒性杂质的相关反应,见表1。
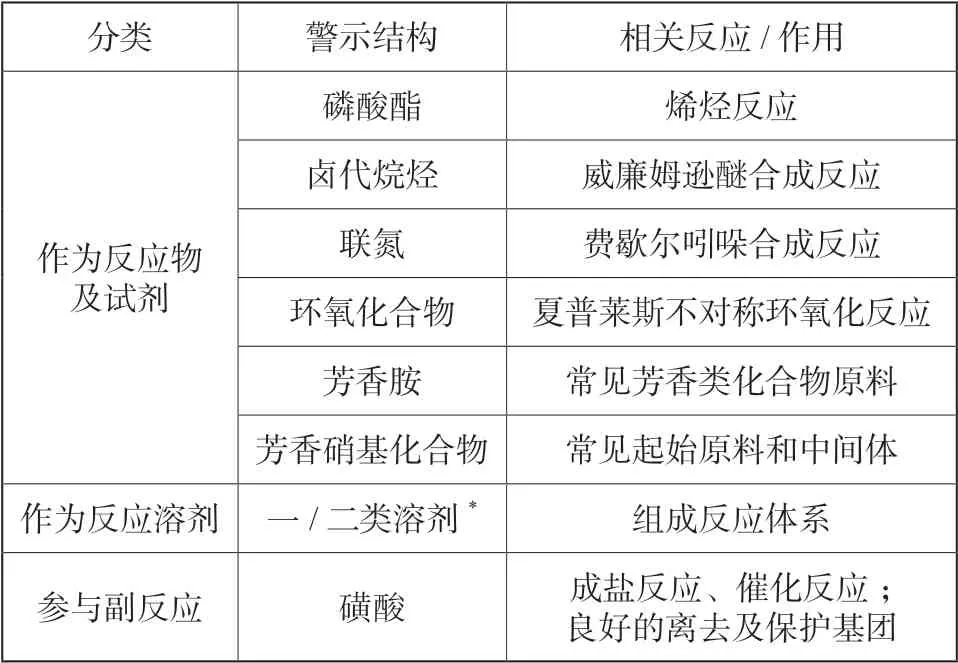
表1 警示结构化合物为原料药引入基因毒性杂质相关反应Tab.1 Structural alerts relevant reactions of genotoxic impurities introducing to drug substances
3 基因毒性杂质分类控制原则及限度要求
3.1 危害性评估
根据杂质的致癌性、基因毒性以及原料药基因毒性,可将杂质分为5 类(表2)[2],并给予不同的控制要求(图2)。当实验数据证实杂质具有致癌性或基因毒性时,首先考虑清除或避免引入该杂质,实际不可行时则采用可接受标准控制杂质含量,常用有允许日暴露量(Permissible Daily Exposures,PDE)及毒性学关注阈值(Threshold of Concern,TTC)两种可接受限度计算方法[2],具体使用见3.2 节。对于无警示结构或警示结构与无基因毒性原料药一致的杂质,则可按照无基因毒性杂质进行控制。若已知原料药具有基因毒性,且杂质具有警示结构或确认基因毒性,则可忽略该杂质带来的致癌风险,也将其列为非基因毒性杂质控制。

表2 基于致癌性及基因毒性归类的5 类杂质Tab.2 5 tier of impurities classified by carcinogenicity and genotoxicity
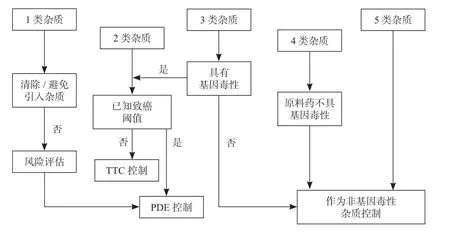
图2 原料药中基因毒性杂质分类控制路径图Fig.2 Control strategies for classified genotoxic impurities in drug substances
3.2 风险表征
1~3 类具有基因毒性的杂质根据可接受摄入量的风险表征原则设计原料药中杂质可接受限度。可接受摄入量是指一个特定的杂质摄入水平,该水平下摄入杂质所增加的致癌风险可忽略不计或低于疾病威胁,若已知杂质的致癌阈值(如1 类杂质及某些3类杂质),可依照该阈值设置PDE,并根据相关治疗周期进行调整。若没有足够的致癌性数据支持杂质限度的设定,则需要采用保守的致癌性估算方法,即TTC 控制。TTC,指未知致癌或其他毒性作用风险的化学物质的可接受摄入量,其以化学物质作用于最敏感物种及最敏感部位能产生50%肿瘤发生率的浓度(TD50)作为计算基准,线性外推至肿瘤发生率百万分之一处而得,在药物研发后期还可将TTC 提高到产生十万分之一肿瘤发生率的浓度,即1.5 μg/d的限度[3]。若药物的暴露短于终生时,满足杂质累计摄入总量不变的前提下还可以放宽其可接受日摄入量,参考表3。

表3 基于TTC 原则及治疗周期的单个基因毒性杂质可接受日摄入量Tab.3 Acceptable daily intake of single genotoxic impurity based on TTC approach and treatment period
4 原料药中基因毒性杂质控制措施
对于基因毒性杂质,总体控制思路是:“避免-清除-控制”[4],即设计合成路线时避开选用基因毒性物料,调整工艺参数及增加纯化手段清除基因毒性杂质,最后开发安全的质量标准及灵敏准确的分析方法以控制残留的基因毒性杂质[5]。根据基因毒性杂质的特性及引入的工艺位置,确定合适的控制手段,详见图3。
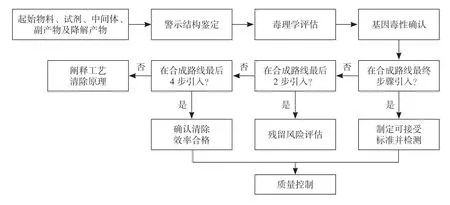
图3 原料药中基因毒性杂质控制流程图Fig.3 Scheme of genotoxic impurities controlling in drug substance
4.1 生产工艺优化
4.1.1 合成路线设计
控制原料药中基因毒性杂质的危害首选措施是避免选用具有基因毒性的原料及试剂,同时选择无基因毒性杂质生产的合成路线。原料药合成过程中常用到磷酸酯、卤代烷烃、烷基磺酸酯、环氧化合物、偶氮、醛类与芳香胺等基因毒性化合物[6-7],其中烷基磺酸酯类及卤代烷烃类化合物还可通过多种副反应生成其他基因毒性杂质,所以应尽可能选择其他化合物代替反应。ICH Q3 规定的1 类溶剂(如:苯、四氯化碳、二氯乙烷)及2 类溶剂(如:氯仿、四氢化萘、己烷、乙二醇)存在致癌风险[8],可按照Q3 指导原则规定的控制方法避免使用或降低至可接受标准以下。
4.1.2 工艺参数调整
然而许多实际情况导致无法避免基因毒性杂质的引入,一些原料的基因毒性警示结构恰好是合成步骤的活性基团,而在研发后期商业化放大后变更合成路线也不现实,这时则需要优化工艺参数以减少基因毒性杂质的含量。通过适当调整工艺中反应pH值、温度、时长及反应溶剂的比例等参数,可以改变基因毒性副产物生成反应的反应程度,达到去除基因毒性杂质的效果。如:磺酸酯具有基因毒性,药物Tasaglitazar[9]生产工艺中使用了芳香类二甲磺酸酯进行醚生成反应,这一步骤向反应体系引入了基因毒性杂质。若将反应pH 值调整为7 并增加回流时间至8 小时,则促进该基因毒性试剂反应完全,反应后期向体系引入水可加速磺酸酯的降解,最终降低杂质残 留。
4.1.3 原料药粗品纯化
原料药生产工艺过程中的回流、萃取、活性炭吸附及重结晶等操作本身就具备去除基因毒性杂质的功能,可以根据杂质及原料药物理化学性质的差异调整参数以清除效率,还可以借助膜过滤、超临界流体萃取及分子印迹吸附等技术进一步纯化原料药[10]。
4.2 质量控制完善
4.2.1 质量标准制定
基于对当前原料药整体特性、生产工艺及物料属性的理解,分析各流程为终产品引入基因毒性杂质的可能性及程度,为杂质检测部分增加合理的基因毒性杂质限度及开发适当的分析方法[11-12]。如果明确存在前述1~3 类基因毒性杂质,应构建专门的控制策略。在连续6 批中试规模或3 批商业规模的原料药中基因毒性杂质检出量低于限度30%的,可将基因毒性杂质检测列为周期性检验项目而不进行常规检测[2]。
4.2.2 原料及过程控制
结合起始物料生产工艺及原料药合成路线中杂质的去向,将起始物料、试剂及中间产物中基因毒性杂质含量控制在原料药限度下,或设置过程控制项目以保证最终产品质量。根据基因毒性杂质引入步骤在工艺中的位置并结合杂质的反应活性、溶解度、解离度及挥发性确定其清除因子,若能合理解释工艺本身对杂质的清除作用,且能明确杂质最终含量低于可接受标准,可不对该杂质进行质量控制[2]。
5 应用
根据上述原料药基因毒性杂质清除及控制的原理,可对符合欧洲标准10.0 版的抗敏感类药物活性成分盐酸西替利嗪的合成路线及生产工艺过程中可能引入的基因毒性杂质的位置及类型进行分析,并根据基因毒性杂质特性提出相应的控制策略。
5.1 盐酸西替利嗪合成路线
盐酸西替利嗪原料药由起始物料4-氯二苯氯甲烷(化合物A)与N-羟乙基哌嗪(化合物B)在碳酸钠溶液中进行亲核取代反应,得到化合物C,其在碱性条件下与氯乙酸钠发生取代反应生成化合物D,后者在两步盐酸反应下最终生成盐酸西替利嗪(化合物F)。反应过程中除溶剂水外,还使用了N,N-二甲基甲酰胺(DMF)以及丙酮等有机溶剂作为反应载体。
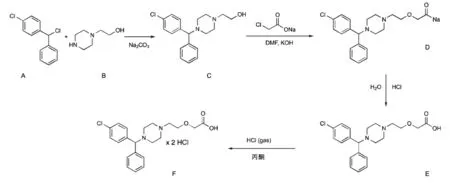
图4 盐酸西替利嗪合成路线Fig.4 Synthesis pathway for cetirizine hydrochloride
5.2 基因毒性杂质筛查
根据基因毒性杂质的来源,结合欧洲药典标准,从起始物料、中间体、残留溶剂、副产物及降解产物等方面对该合成路线生产的盐酸西替利嗪的潜在基因毒性杂质进行分析。
5.2.1 药典杂质
表4 汇总了欧洲药典10.0 版中记载的盐酸西替利嗪生产过程中可能出现的杂质及其可能来源,可将药典杂质作为成品常规必检项目,同时根据实际合成路线及工艺条件,增加包括工艺相关的有关物质及残留溶剂检测项目。
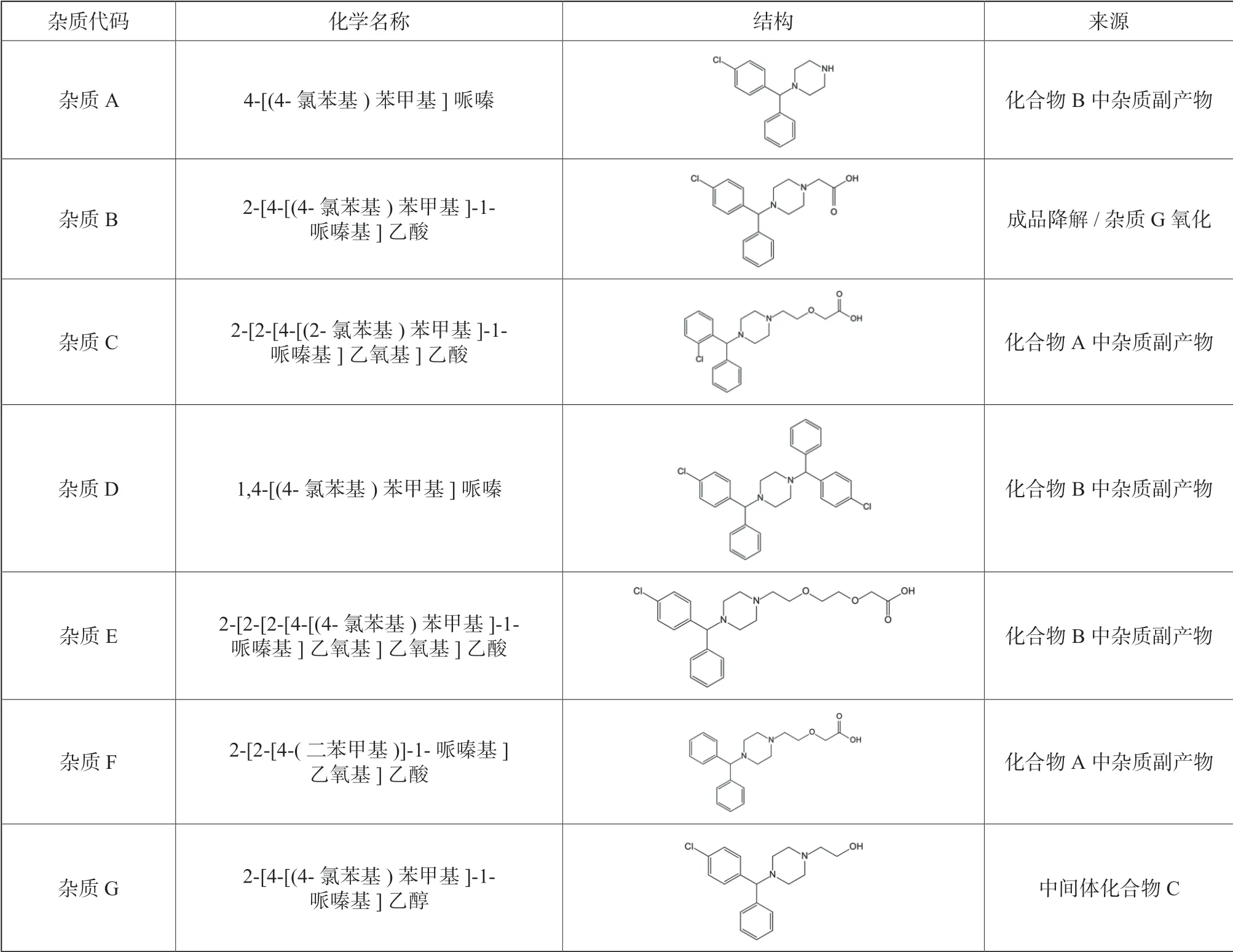
表4 盐酸西替利嗪欧洲药典10.0 版记载杂质信息汇总Tab.4 Information of impurities of cetirizine hydrochloride in European Pharmacopeia 10.0
5.2.2 起始物料及其副产物
该合成路线包含两个起始物料(化合物A、B),其中化合物A 为卤代烃,具有基因毒性警示结构,需要纳入后期基因毒性杂质控制范畴。而起始物料化合物A 与B 本身均可能携带相应杂质,在第一步亲核取代反应中起始物料引入的杂质能通过副反应产生杂质A、C、D、E 和F,经过比对,未在这五类杂质中发现基因毒性警示结构,故可按照药典标准以普通杂质进行控制。
5.2.3 中间体
该合成路线包含三个中间体(化合物C、D、E),三个中间体化学结构均不具有基因毒性警示结构,化合物D 在酸性条件下不稳定,在后续反应中将转化为化合物E,且化合物E 与成品具有相同化学结构,故可按照药典标准仅对化合物C 以普通杂质进行控 制。
5.2.4 残留溶剂及其副产物
该合成路线在第二步亲核取代反应中使用到了ICH Q3C 规定[13]的二类溶剂DMF,具有一定的基因毒性,需按照指导原则对其进行中间控制及放行检查。同时关注DMF 及丙酮在反应过程中产生的潜在基因毒性副产物。如DMF 曾被报道[14]在酸性条件下不稳定,容易生成基因毒性杂质N-二甲基亚硝胺(NDMA);丙酮在酸性条件下易与自身发生反应(见图5),产生副产物2,6-二甲基-2,5-庚二烯-4-酮(化合物H)、3-氯-4-甲基-2-戊酮(化合物J)、4-氯,甲基-2-戊酮(化合物K)和2,6-二氯,二甲基-4-庚酮(化合物L),四个副产物分别具有烯酮及卤代烃等基因毒性警示结构,需要纳入基因毒性杂质控制范畴。
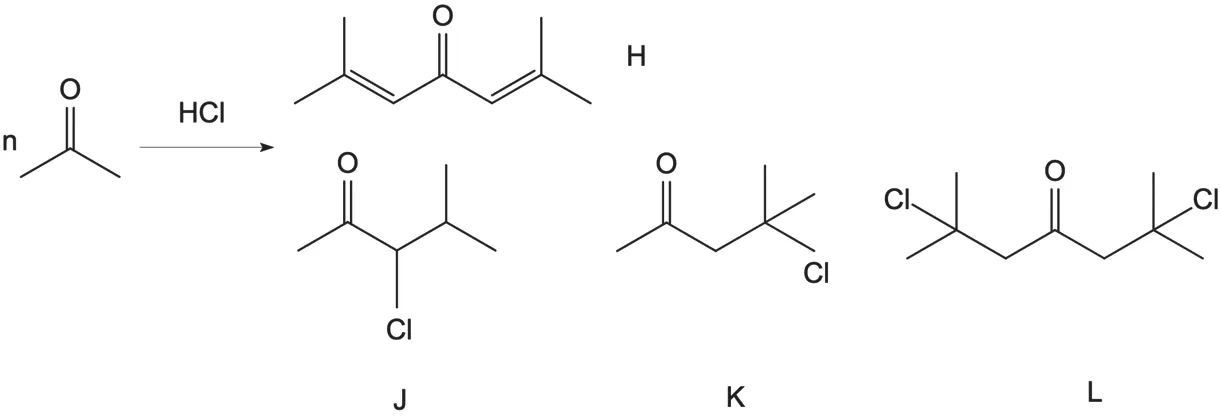
图5 丙醇在酸性条件下所产生的副产物Fig.5 By-products acetone introduced in acid condition
5.2.5 降解产物
成品及杂质G 在存储运输过程中容易降解或氧化生成杂质B,因其不具有基因毒性警示结构,故可按照药典标准以普通杂质对杂质B 进行放行检测、复验期检测及稳定性检测。
5.3 基因毒性杂质控制
根据上述基因毒性杂质筛查结果,需要按照基因杂质清除及控制策略对起始物料化合物A;残留溶剂DMF;副产物NDMA 及副产物H~L 等进行严格控制。
对于起始物料A,由于其的引入位于四步合成反应的第一步,距离成品盐酸西替利嗪的成盐及后续结晶纯化步骤较远,且其低溶解度的性质使其在该步结晶过程中与可溶性中间体C 分离,难以参与后续反应,故可采用中间控制的手段控制其含量。如在第一步亲核取代后将其作为杂质项目,控制含量限度在0.1%以下即可保证其后续清除效率,若连续三批验证批次检测结果均低于标准30%,也可不纳入常规检验。
对于残留溶剂DMF,由于其归属于具有基因毒性的二类溶剂范畴,且其在后续反应的酸性条件下不稳定易生成NDMA,故需要在该步骤设置严格的中间控制,同时在成品质量标准中进行把关,可根据ICH Q3C 将控制限度设置为8.80×10-4以保证DMF的清除同时预防其在后续酸性反应中生成NDMA。
溶剂丙酮在酸性条件下生成基因毒性副产物H~L,但目前尚未找到合适的替代溶剂,故需要通过工艺参数调整、多步纯化以及质量控制等手段保证成品中该类杂质的含量控制在可接受水平之中。副反应效率与保温回流时间以及反应pH 值相关,可在工艺验证数据支持下适当缩短保温回流时间,少量多次加入盐酸以保持pH 值变化稳定,降低整体副反应效率,减少副产物生成;化合物H~L 具有良好的醇溶性,可利用乙醇对盐酸西替利嗪粗品进行重结晶纯化;同时根据TTC 原则计算杂质限度:目前尚未有文献记载化合物H~L 的致癌剂量,考虑到盐酸西替利嗪是过敏患者长期用药的选择,剂量为20 mg/ d,以基因毒性杂质可接受日剂量1.5 μg/d 作为计算依据,故基因毒性副产物H~L 的可接受标准应设置为7.5×10-4,质量控制需要开发准确的分析方法,通常使用到HPLC-MS 或LC-MS 等检测手段。
6 结束语
随着对原料药合成过程中基因毒性杂质的引入认知的加深,我国原料药行业改变了过往对基因毒性杂质控制的“检出”模式,逐步转型为“质量源于设计”模式。通过潜在基因毒性杂质危害性评估确定杂质分类及控制策略,进而优化原料药的合成路线及生产工艺,建立更加合理的质量标准及质量控制体系,从源头降低原料药中基因毒性杂质的引入,并通过合适的清除手段降低杂质含量,最终保障用药安全。然而基因毒性杂质控制仍存在诸多难点,首先要求企业对原料药合成路径及反应机理有深刻理解,其次需要建立痕量水平分析方法,最后痕量杂质清除要求采用更先进的纯化手法,无疑对我国药品研发企业提出了新的挑战。
