遗传性痉挛性截瘫IBA57基因新发突变1例
2021-06-27孙艳美张宁罗艳李亚丽
孙艳美,张宁,罗艳,李亚丽
(河北省人民医院生殖遗传科,石家庄 050051)
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)是一种罕见的、具有高度临床和遗传异质性的遗传性神经系统退行性病变。HSP遗传基础复杂,涉及孟德尔遗传模式(包括常染色体显性遗传、常染色体隐性遗传和X连锁隐性遗传)和线粒体母系遗传,其中以常染色体显性遗传最为常见。最常见的病理特征是皮质脊髓束和后柱长下行运动神经纤维远端轴索病变。HSP发病机制迄今尚不清楚,目前多认为与膜囊泡运输、轴突运输、脂质代谢、细胞器形态发生和分布、线粒体功能和髓鞘化过程等受到影响有关[1-2]。HSP常合并多系统病变,临床表型也多种多样,误诊和漏诊率高。本文报道了1例HSP病例,并检测到IBA57基因新发突变。
1 材料与方法
1.1 临床资料
孕妇34岁,孕3产2,分别于2007、2015年足月阴道分娩两子,两子均发生走路不稳伴视力下降。现再次妊娠,早孕期超声显示双胎(双绒双羊),要求明确两子所患疾病性质,并对本次妊娠胎儿行产前诊断。夫妇双方身体健康、非近亲结婚,孕期及孕前期均无毒物、放射线接触史,双方家族中均无类似疾病史或其他遗传病史。家系图见图1(箭头所指为先证者)。
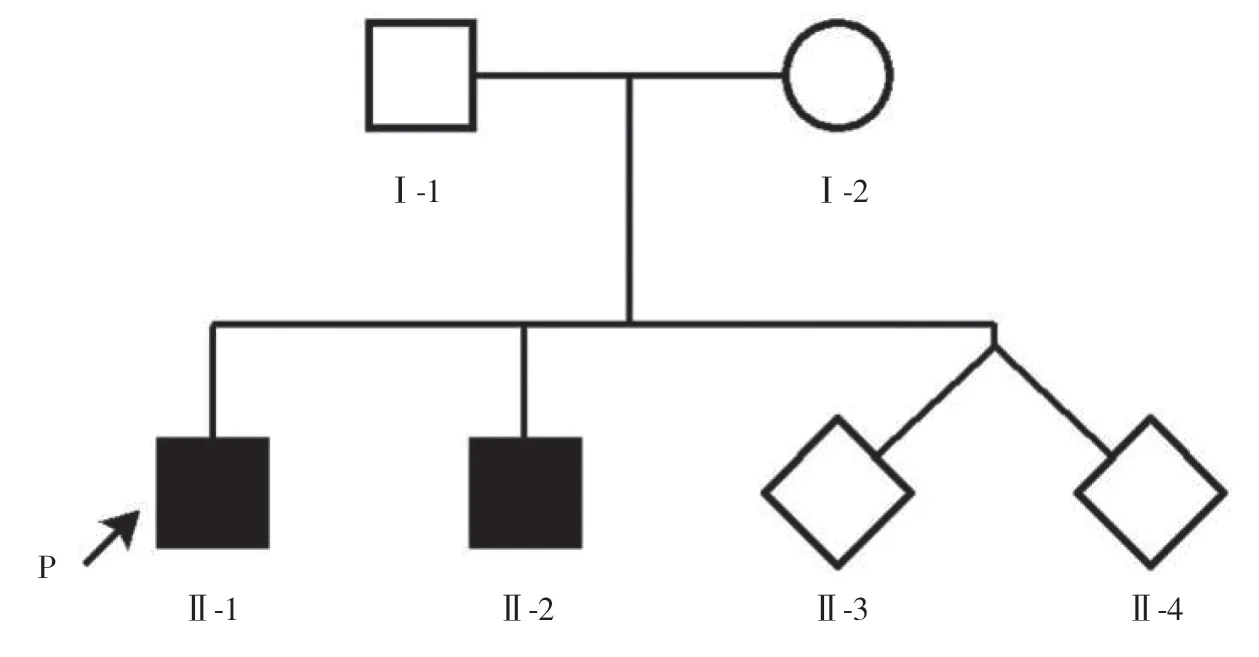
图1 家系系谱图
先证者(Ⅱ-1)3岁时出现视力严重下降,有光感,眼科检查示视神经萎缩,经治疗3年后视力恢复至0.3~0.4,7岁时出现双下肢肌张力异常,走路不稳,神经系统检查提示肌力增高,初步诊断为“截瘫”,给予康复治疗后略好转,出现发热或上呼吸道感染后症状加重。二子(Ⅱ-2)自1岁余开始出现视力下降,有光感,眼科检查示视神经萎缩,5岁出现走路不稳,容易跌倒,未给予特殊治疗,冬季症状加重,病情逐年进展。
1.2 研究方法
抽取先证者(Ⅱ-1)外周血行全外显子测序,并抽取二子(Ⅱ-2)、夫妇双方外周血,提取DNA,对可疑致病性变异行Sanger测序验证。孕妇于孕17周行羊膜腔穿刺,抽取15 mL羊水行Sanger测序验证。孕妇预产期年龄近35岁,经孕妇本人及家属同意,同时抽取20 mL羊水行胎儿染色体核型分析。
采用天根DNA试剂盒提取外周静脉血基因组DNA及羊水DNA。全外显子高通量测序采用Illumina Hiseq 2000高通量测序仪(美国Illumina公司),Sanger测序采用ABI3730xl DNA测序仪(美国赛默飞公司)。染色体核型分析采用Giemsa染色,Leica全自动染色体扫描平台(德国Leica公司)分别从2个培养体系中采集、计数核型。
2 结果
2.1 先证者基因测序结果及疾病诊断
二代测序显示先证者(Ⅱ-1)患儿IBA57基因存在c.22C>T(p.R8X)和c.341G>C(p.G114A)位 点杂合突变,IBA57基因c.341G>C(p.G114A)杂合突变为母源,IBA57基因c.22C>T(p.R8X)杂合突变为父源,二子(Ⅱ-2)同时携带IBA57基因c.22C>T(p.R8X)和c.341G>C(p.G114A)位点(图2、3),为复合杂合子。结合患儿表型及基因突变位点,考虑诊断为HSP 74型。
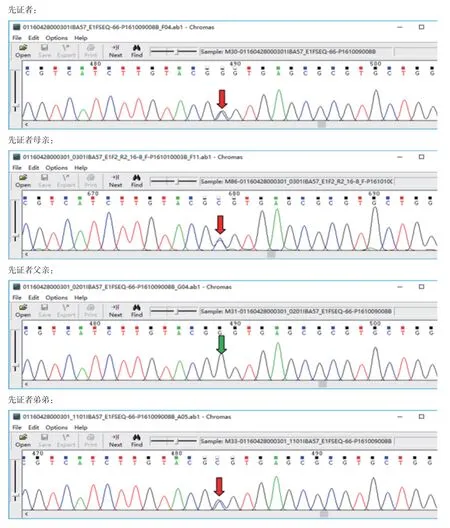
图3 IBA57基因c.341G>C(p.G114A)突变位点
2.2 胎儿产前诊断
孕妇于孕17周行羊膜腔穿刺产前诊断,提取羊水细胞DNA行Sanger测序,基因检测结果显示:一胎儿未携带IBA57基因c.22C>T(p.R8X)和c.341G>C(p.G114A)突变位点(图4),一胎儿携带IBA57基因c.341G>C(p.G114A)突变位点(图5)。羊水染色体核型分析示两胎儿均为正常核型。

图4 F1胎儿IBA57基因突变位点
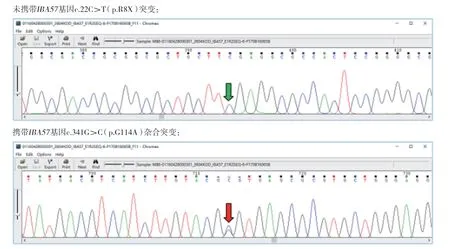
图5 F2胎儿IBA57基因突变位点
3 讨论
HSP是一种神经系统退行性病变或神经发育障碍性疾病综合征,主要的神经系统症状和体征为下肢痉挛和无力[1]。其临床表型复杂,分型众多,存在明显的遗传异质性,给临床实践带来了极大的挑战。HSP的诊断多结合临床表型、家族史及基因测序结果,目前认为基因测序是诊断HSP的金标准。神经系统检查显示皮质脊髓束受累,例如痉挛、反射亢进,但要注意与其他原因引起的痉挛性截瘫鉴别,如脊髓损伤[3];合并其他系统症状的复杂性痉挛性截瘫,可同时表现出其他神经系统或非神经系统体征,如视神经病变、共济失调、癫痫、周围神经病变及椎体外系病变等,且其他系统的症状可早于痉挛性截瘫发作之前出现[4-6]。痉挛性截瘫患者肌电图可呈现神经源性损伤,磁共振检查可呈现是颈髓、胸髓及胼胝体变薄、脑白质病变等[7-8]。因此,临床医生和临床遗传学家必须充分了解痉挛性截瘫的临床表型、神经影像学特征和遗传学特点,以提供准确的遗传学评估和咨询。
本研究中,致病基因为IBA57基因c.22C>T(p.R8X)和c.341G>C(p.G114A)杂合突变。在先证者(Ⅱ-1)中发现的IBA57基因c.22C>T(p.R8X)杂合突变可导致生物多肽链合成提前终止,属于无义突变,理论上具有致病性,但是该位点仅在空泡性脑白质病中报道过[9],痉挛性截瘫病例未见报道;c.341G>C(p.G114A)位点突变引起多肽链合成时丙氨酸替换为甘氨酸,该突变目前未见文献报道,生物信息分析软件Mutation_Taster预测为“有害”,SIFT及PolyPhen2预测为“良性”。传递分析表明,先证者父亲携带IBA57基因c.22C>T(p.R8X)杂合突变,先证者母亲携带IBA57基因c.341G>C(p.G114A)杂合突变,先证者弟弟(Ⅱ-2)同时携带上述2个突变,属复合杂合子。综合以上遗传学特点及临床表型,确定其为致病基因并以常染色体隐性方式遗传,该家系所患痉挛性截瘫为HSP 74型。
HSP 74型是一种以常染色体隐性遗传的临床综合征,由IBA57基因的位点突变导致,多为纯合型。IBA57编码的蛋白参与血红蛋白合成中线粒体的铁硫簇合成。HSP 74型多在10岁前起病,典型的临床表现为缓慢进行性下肢痉挛性截瘫、轴突运动性周围神经病变、视神经萎缩、视力低下和视野缺损等[10]。目前,针对该病尚无有效的治疗方法,临床上多采用对症治疗,如给予巴氯芬或替扎尼定抗痉挛治疗,通过步态训练或矫形器改善运动障碍情况,提高平衡能力,且这种治疗在成年之前较成年之后效果好[11-13]。目前针对该病的治疗研究较少,还需进一步推进更多治疗方案,提高治疗效果。
单基因病产前诊断是防治出生缺陷的重要方法,对降低出生缺陷率,减少家庭及社会负担意义重大。本研究立足于出生缺陷二级预防,借助于基因测序进行了产前遗传学诊断,旨在为此类疾病的产前诊断提供参考资料。
