葡萄糖氧化酶在毕赤酵母中的高效分泌表达
2021-06-21魏东升段广东钱江潮
魏东升, 段广东, 钱江潮
(华东理工大学生物反应器国家重点实验室,上海 200237)
葡萄糖氧化酶(GOD,EC 1.1.3.4)是一种氧化还原酶,由两个亚基组成同源二聚体,不同来源的GOD理化性质存在差异[1],分子量在130~175 kDa范围内的GOD可催化β-D-葡萄糖发生氧化反应生成葡萄糖酸和过氧化氢[2]。GOD被广泛应用于食品、化工、生物医疗等领域。在食品行业,GOD可被用于催化葡萄糖耗尽真空袋中氧气,抑制微生物生长和繁殖,延长食品保质期[3],还可提升面制品的口感[4]。在化工领域,GOD不仅常被应用于漂白脱色工艺[5],还是葡萄糖酸及其衍生物生产中的关键酶。医疗领域中,GOD是葡萄糖检测试剂盒的关键原料[6],也被加入牙膏中用于降低口腔疾病发生率[7],还能作为生物电池的电极,为生物传感器和人造器官提供持续的能源[8]。因此,大规模高效生产GOD具有重要的经济价值。
目前工业水平的GOD生产,主要以黑曲霉和青霉作为生产菌株,但存在酶活水平低、分离纯化复杂的问题。为了实现GOD的高效生产,研究者已利用不同宿主尝试了GOD的高效异源表达。但是,大肠杆菌(Escherichia coli)异源表达GOD时,容易形成无活性的包涵体[9]。酿酒酵母表达的GOD,由于过糖基化的原因,会降低重组GOD与葡萄糖的亲和力和催化效率[10]。而毕赤酵母(Pichia pastoris)表达的GOD并不会产生过糖基化修饰的现象。Yamaguchi等[11]在P. pastoris中实现了GOD的分泌表达,但酶活只有1.23 U/mL。Gao等[12]通过密码子优化,使GOD酶活达到了615 U/mL。顾磊等[13]通过对GOD进行定点突变以及改造P. pastoris的翻译加工和转运的过程,提高了GOD的表达能力,在3 L发酵罐中,GOD分泌产量达到1 972 U/mL,为目前文献报道的最高水平。
P. pastoris遗传背景清楚,易于进行遗传操作,分泌的蛋白糖基化适中,是一种进行外源蛋白表达的常用宿主。尤其是利用P. pastoris进行蛋白分泌表达,可大大简化分离纯化的过程,具有重要的应用价值。但是,外源蛋白的高效表达会对重组菌的内质网产生胁迫压力,影响外源蛋白的折叠和转运。因此,共表达辅助折叠因子,提高内质网处理蛋白的能力成为促进外源蛋白分泌表达的常用策略。例如,HAC1是由HAC1基因编码的一个参与未折叠蛋白反应(Unfolded Protein Response, UPR)的转录因子,共表达HAC1可提高GOD和白介素等不同外源蛋白在P. pastoris中的分泌表达水平[13-14]。此外,外源蛋白的表达也会对宿主的碳代谢产生影响,Nocon等[15]通过强化磷酸戊糖途径的SOL3和三羧酸循环的MDH1基因,使得重组超氧化物歧化酶产量提高了40%。
为了构建高效分泌表达GOD的P. pastoris,在本文中,黑曲霉来源的GOD序列被优化后用于构建分泌表达GOD的P. pastoris重组菌,通过增加抗生素浓度,筛选得到GOD高拷贝重组菌株。在此基础上,进一步共表达多个辅助折叠因子以及强化碳代谢途径,探索多种组合策略组合对GOD分泌表达影响,获得了第2代GOD高产菌,并在5 L和50 L发酵罐中对高产菌的工业应用前景进行考察和验证。
1 材料和方法
1.1 材料
1.1.1 菌株和质粒 大肠杆菌DH5α购自TaKaRa公司,毕赤酵母GS115购自Invitrogen公司;pPIC9K:购自Invitrogen,含有α-MF信号肽,以AOX1为启动子,并能通过同源基因his整合到P. pastoris基因组上的表达载体(AmpR);pPIC9K-GOD:本课题构建,以pPIC9K为载体,分泌表达GOD的重组质粒(AmpR);T-GAPDH:本课题构建,将GAPDH基因片段整合到pUCm-T载体(含有LacZ基因,用于克隆含有A末端的线性载体)上;pAOX-SSN:本实验室保存,包含P.pastoris染色体ADRH和DDKC基因同源序列,以AOX1调控基因表达的载体(ZeocinR)[16]; pAOX-PDI1:本实验室保存,以pAOX-SSN为载体所构建AOX1调控PDI1表达的载体(ZeocinR)[16]; pAOX-MPD1:本实验室保存,以pAOX-SSN为载体所构建AOX1调控MPD1表达的载体(ZeocinR)[16]; pAOX-PDI2:本实验室保存, 以pAOX-SSN为载体所构建AOX1调控PDI2表达的载体(ZeocinR)[16]; pAOX-BIP:本实验室保存,以pAOX-SSN为载体所构建AOX1调控Bip表达的载体(ZeocinR)[16]; pAOX-SIL1: 本实验室保存,以pAOX-SSN为载体所构建AOX1调控SIL1表达的载体(ZeocinR)[16]; pAOX-HAC1:本实验室保存,以pAOX-SSN为载体所构建AOX1调控HAC1基因表达的载体(ZeocinR)[16]; pAOX-ZWF1: 本实验室保存,以pAOX-SSN为载体所构建AOX1调控ZWF1表达的载体(ZeocinR)[17]; pAOX-SOL3: 本实验室保存,以pAOX-SSN为载体所构建AOX1调控SOL3表达的载体(ZeocinR)[17]; pAOX-MDH1: 本实验室保存,以pAOX-SSN为载体所构建AOX1调控MDH1表达的载体(ZeocinR)[17]; pAOX-GDH3: 本实验室保存,以pAOX-SSN为载体所构建AOX1调控GDH3表达的载体(ZeocinR)[17]。
1.1.2 酶和试剂 工具酶,包括T4连接酶、限制性内切酶(BamH I、EcoR I、XhoI、NotI、SalI等)、标准蛋白分子量Marker、标准核酸分子量Marker均购自大连TaKaRa生物技术有限公司;PCR所用的rTaqPCR聚合酶和pfuPCR 聚合酶均购自北京天根生化科技有限公司。
所用分子克隆试剂盒,包括质粒抽提试剂盒、DNA凝胶回收试剂盒、核酸纯化试剂盒以及酵母基因组DNA提取试剂盒,均购自杭州爱思进生物技术有限公司。
1.2 方法
1.2.1 表达载体的构建 文中所用的引物见表1(划线部分为限制性酶切位点)。构建AOX1启动子调控GOD表达的质粒pPIC9K-GOD时,根据黑曲霉的GOD蛋白序列(UniProt: P13006),按照毕赤酵母的密码子偏好性进行优化后,合成Pgod基因,使用引物GODF和GODR扩增GOD序列,通过EcoR I和NotI酶切位点连接到pPIC9K载体上,构建GOD分泌表达载体pPIC9K-GOD。

表1 本研究中所使用的引物Table 1 Primers used in this study
1.2.2 重组菌的构建和筛选 用限制性内切酶SalI线性化质粒pPIC9K-GOD,纯化后通过电击毕赤酵母GS115感受态细胞,利用GS115的组氨酸缺陷型特征,在MD(Minimal Dextrose)固体培养基上筛选重组菌,并经PCR和测序验证。
使用无菌水洗涤收集MD培养皿中的重组菌,涂布在含有不同质量浓度(0、0.25、0.5、0.75、1.0、1.5、1.75、2.0 mg/mL)遗传霉素G418的YPD(Yeast Peptone Dextrose)培养皿上。挑取耐高抗生素浓度的重组菌,考察GOD产量并测定高产菌的拷贝子数。
获得高拷贝高产菌G/GODM后,为了进一步共表达折叠因子或碳代谢途径基因,将pAOX-PDI1、pAOX-MPD1、pAOX-PDI2、pAOX-BIP、pAOX-SIL1、pAOX-HAC1、pAOX-ZWF1、pAOX-SOL3、pAOXMDH1和pAOX-GDH3经XhoⅠ/EcoRⅠ双酶切后得到线性化片段,电击转化G/GODM,利用共表达基因两端所插入的ADRH基因和DDKC基因同源序列,通过同源重组的方式分别整合到菌株G/GODM的基因组上。经过博来霉素抗性筛选得到阳性克隆,经PCR验证及测序后筛选到正确的重组菌。
1.2.3 拷贝子数目测定 按照文献已报道的方法[18]测定重组菌中GOD基因拷贝数。将参比质粒T-GAPDH、pPIC9K-GOD分别稀释至每微升中有109、108、107、106、105、104、103个质粒、分别取1 μL上述质粒稀释液作为模版进行荧光定量PCR,以Ct值(循环次数)为纵坐标、起始模版质粒浓度的对数值为横坐标,绘制标准曲线。
以重组菌的基因组DNA为模板,以引物gapdhF、gapdhR、qGODF和qGODR进行荧光定量PCR。根据测得的Ct值和标准曲线确定重组菌GAPDH和GOD的起始模版量,计算两者起始模版数量的比值,得到重组菌中GOD基因的拷贝子数。
1.2.4 重组菌的培养方法 在摇瓶中进行培养时,将重组菌接种到含25 mL的BMGY(Buffered Glycerol-Complex Medium)培养基中,于30 ℃、220 r/min培养约18 h至OD600在4~6之间,收集菌液至无菌的50 mL离心管中,于4 ℃、4 000 r/min离心5 min,用BMMY(Buffered Methanol-Complex Medium)培养基重悬菌体,并调节OD600在1左右,后转移至装有50 mL BMMY培养基的500 mL摇瓶中进行诱导培养,每隔24 h取样,并补充1%(体积分数)的甲醇。
发酵罐中的培养分为3个阶段:分批发酵阶段、甘油流加阶段和甲醇诱导阶段。在分批发酵阶段,控制通气比约为1.5 VVM,搅拌转速为600 r/min,待分批发酵结束、溶氧(Dissolved oxygen, DO)开始反弹时,开始补加甘油,通过控制甘油流加速率,使DO维持在5%~10%之间,当OD600达到400时,停止补加甘油,待DO反弹至100%,饥饿培养30~60 min后,开始缓慢流加甲醇,通过调整甲醇流加速率、搅拌转速、通气量等操作变量,控制DO在0~5%之间。整个培养过程中,通过补加氨水维持pH值在5.5,批发酵和甘油流加阶段温度设定在30 ℃,诱导阶段温度设定在22 ℃。
1.2.5 GOD活性测定 GOD酶活定义:在37 ℃下,每分钟催化1 μmol的β-D-葡萄糖生成葡萄糖酸的酶量定义为1个单位酶活U。采用终点法测定GOD酶活:在10 mL离心管中依次加入2.5 mL的0.21 mmol/L的邻联茴香胺、0.3 mL的0.18 g/mL葡萄糖、0.1 mL的90 U/mL的辣根过氧化物酶,37 ℃保温5 min后,向离心管中加入酶液,反应3 min后,加入2 mol/L硫酸终止反应,取出离心管,测定OD500的吸光值。
将GOD标准品稀释至0.5、1.0、1.5、2.0、2.5、3.0 U/mL,按照GOD酶活测定方法,测定反应3 min后的OD500值,以OD500值为横坐标,以GOD酶活为纵坐标,绘制标准曲线用于计算酶活(Activity):Activity=(0.157 8×OD500−0.003 3)×V/V0,其中V为反应总体积,V0为加入酶液的体积。
2 实验结果
2.1 构建GOD高产菌G/GODM
为了实现GOD在P. pastoris中的分泌表达,选择了pPIC9K载体,在EcoRI和NotI酶切位点插入Pgod基因,构建GOD分泌表达载体pPIC9K-GOD(图1(a)),经SalI酶切线性化后电击转化毕赤酵母GS115,在MD固体培养基上挑取阳性单菌落,使用引物GODF/GODR进行菌落PCR验证重组菌,并经测序验证重组菌G/GOD构建成功(图1(b))。

图1 酶切验证质粒pPIC9K-GOD(a)和重组菌G/GOD、G/GODM的PCR验证(b)Fig. 1 Enzyme digestion to confirm plasmid pPIC9K-GOD (a)and verification of recombinant strain G/GOD and G/GODM by PCR analysis (b)
在线性化pPIC9K-GOD转化P.pastorisGS115的MD培养板上,用无菌水洗涤收集阳性重组菌,涂布在含有不同质量浓度(0、0.25、0.5、0.75、1.0、1.5、1.75、2.0 mg/mL)遗传霉素G418的YPD培养皿上,获得多株耐G418质量浓度达到2.0 mg/mL的重组菌,在摇瓶中初步筛选后,挑选得到产量最高的菌株G/GODM(图1(b))。测定G/GOD和G/GODM的拷贝子数目分别为1和7。
在摇瓶中培养重组菌G/GOD和G/GODM,并与整合了空载质粒pPIC9K的对照菌G/9K相比,重组菌G/GOD和G/GODM的生长速率未发生改变(图2(a) ),表明外源蛋白GOD表达并没有影响重组菌生长,拷贝子数目的增加也对菌株生长没有影响。在对照菌G/9K中无法检测到GOD酶活,而两株重组菌中的酶活在加入甲醇诱导后开始升高,但多拷贝重组菌G/GODM胞外产酶远高于单拷贝菌G/GOD。G/GOD的胞外GOD产量在144 h达到最高,单位细胞干重酶活为714.2 U/g。G/GODM的胞外单位菌体酶活在120 h达到最高,为5 843.2 U/g(图2(b)),是G/GOD最高产量的8.2倍。因此,在P. pastoris中整合高拷贝Pgod基因可得到GOD高产菌G/GODM。
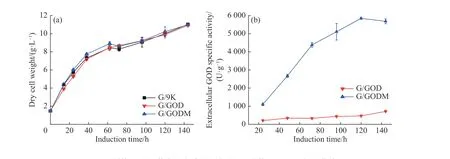
图2 不同拷贝重组菌在摇瓶中的生长(a)和胞外GOD比活(b)曲线Fig. 2 Growth (a) and extracellular GOD specific activity (b) curve of the recombinant strains containing different copy of Pgod gene in shake flask
2.2 强化蛋白折叠分泌和碳代谢途径提高G/GODM的GOD产量
在得到GOD高产菌G/GODM后,为了进一步提高GOD的产量,通过强化G/GODM的蛋白折叠分泌碳和碳代谢途径,尝试构建第2代高产重组菌,结果如图3所示。基于本实验室的前期工作[16-17],选择了P. pastoris分泌折叠途径的基因PDI1、MPD1、PDI2、Bip、SIL1、HAC1,以及碳代谢途径基因ZWF1、SOL3、MDH1、GDH3,并利用前期构建的相应表达载体pAOX-X(X代表以上各基因),通过同源重组的方式整合到重组菌G/GODM基因组的RH-DDKC位点,即两个相邻基因ADRH(PAS_chr1-4_0191)和DDKC(PAS_chr1-4_0192)处。
挑取转化得到的重组菌,分别用引物RHF/AOXR和CYCTTF/DDKCR验证目的片段在上下同源臂位点的整合(如图4),分别获得了强化PDI1、MPD1、PDI2、Bip、SIL1、HAC1、ZWF1、SOL3、MDH1、GDH3基因的重组菌G/GMP1、G/GMMP1、G/GMP2、G/GMB、G/GMS1、G/GMH1、G/GMZ1、G/GMS3、G/GMMD1和G/GMG3。

图3 二代重组菌的构建Fig. 3 Construction of second-generation recombinant strains
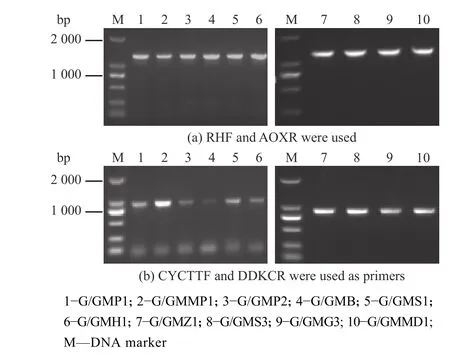
图4 二代重组菌的验证Fig. 4 Verification of second-generation recombinant strains
在摇瓶中培养重组菌,测定不同重组菌的胞内外酶活,结果如图5所示。由图可见,各重组菌都可有效将GOD分泌至胞外,诱导120 h、强化HAC1和PDI1后,与出发菌G/GODM相比,重组菌G/GMH1和G/GMP1的胞外单位菌体酶活分别提高了54.4%和32.7%,达到9 022.1 U/g和7 753.6 U/g;强化PDI2、SOL3和MDH1表达后重组菌胞外单位菌体酶活分别提高了8.9%、6.3%和11.6%。其余基因的共表达均未促进GOD胞外产量,其中共表达MPD1对产量的影响最大,G/GMMP1胞外单位菌体酶活只有2 330.6 U/g,比G/GODM下降了60%。从各重组菌的胞内酶产量来看,共表达这些基因的影响不大。
选择GOD分泌产量提高的5个重组菌,考察它们的生长(图6(a))和胞外酶活(图6(b))。在这5个菌中,只有G/GMH1的生长速度低于出发菌G/GODM。从胞外GOD产量来看,除G/GMS3外,其他重组菌均明显高于G/GODM,其中G/GMH1的胞外酶活最高,在120 h达到最高(9 022.1 U/g),比G/GODM的最高酶活提高了54.4%。
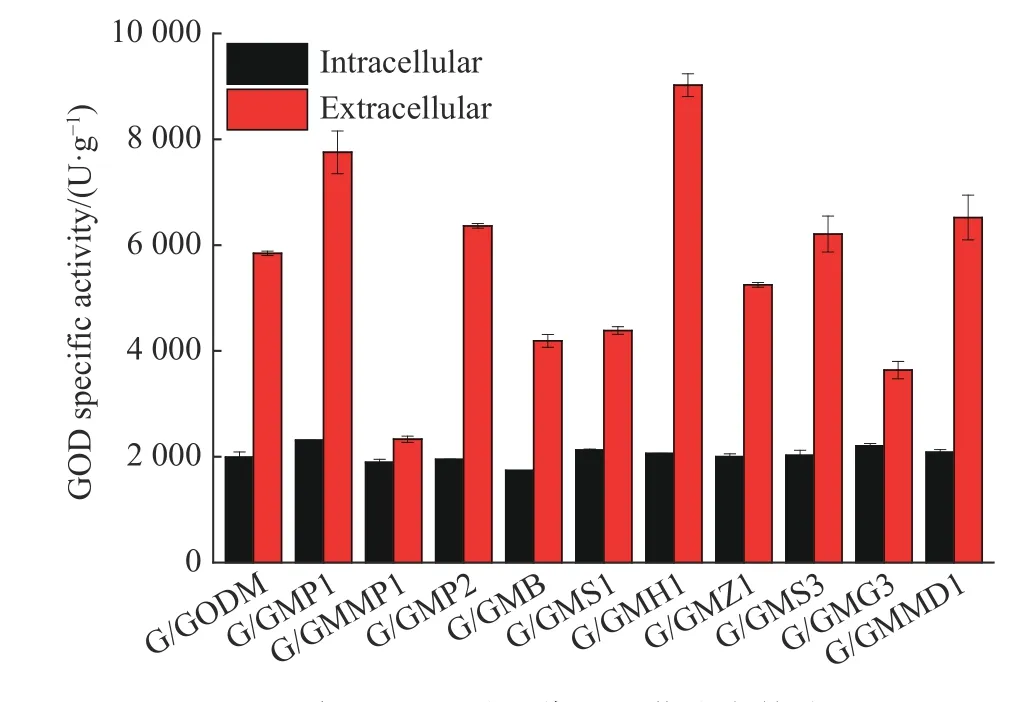
图5 诱导120 h时二代重组菌胞内外酶活Fig. 5 Extracellular and intracellular GOD specific activity of the second-generation recombinant strains after 120 h of induction
2.3 反应器中考察高产重组菌的特性
根据摇瓶培养得到的结果,挑选高产重组菌G/GODM以及两株第2代高产菌G/GMP1和G/GMH1,以单拷贝菌G/GOD为对照,在5 L反应罐中比较它们的生长曲线和产酶能力,结果如图7所示。如图7(a)所示,在甲醇诱导阶段,G/GMH1的产菌量略低于其余3株重组菌,与摇瓶结果一致。各重组菌在诱导阶段持续分泌GOD,随着诱导培养的进行,胞外酶产量不断升高(图7(b)),诱导144 h后,单拷贝菌G/GOD胞外酶产量最低,重组菌G/GMH1的胞外酶产量最高,单位菌体酶活最高达到7 030.4 U/g,是G/GOD的6.6倍。除了G/GOD外,其余3株重组菌的胞外酶产量均低于摇瓶水平,G/GODM和G/GMP1尤其显著。诱导结束时,G/GODM、G/GMP1、G/GMH1的酶活分别达到377.7、490.9、1 010.5 U/mL,分别是单拷贝菌G/GOD的2.2、2.8、5.8倍。

图6 二代高产重组菌的生长(a)和胞外酶活(b)Fig. 6 Growth (a) and extracellular GOD specific activity (b) of the second-generation recombinant strains
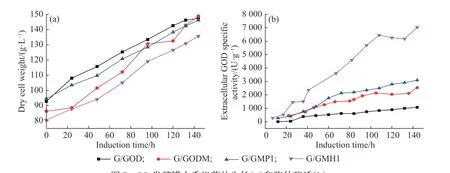
图7 5 L发酵罐中重组菌的生长(a)和胞外酶活(b)Fig. 7 Cell growth (a) and extracellular GOD specific activity (b) of recombinant strains in the 5 L bioreactor
对于胞外酶产量最高的G/GMH1,进一步在50 L发酵罐考察其特性。培养24 h时开始流加甲醇诱导,此后DO一直保持较低水平,而摄氧率(Oxygen Uptake Rate,OUR)和二氧化碳释放率(Carbon Dioxide Evolution Rate,CER)都在快速上升至48 h后基本稳定,此后呼吸熵(Respiratory Quotient,RQ)也相对稳定(图8(a))。随着甲醇诱导的开始,重组菌持续向胞外分泌GOD(图7(b)),至发酵结束时,单位菌体和体积酶活均达到最高,分别为6 656.6 U/g和1 150.5 U/mL。这一结果表明,G/GMH1可在50 L发酵罐中实现较高的GOD分泌表达,具有良好的工业应用前景。

图8 G/GMH1在50 L发酵罐中分批发酵的胞外GOD酶活、细胞干重、DO、OUR、CER、RQ时间曲线(a)和培养上清液的蛋白电泳(b)Fig. 8 Time courses of extracellular GOD specific activity, Dry cell weight, DO, OUR, CER, RQ (a) and protein electrophoresis of supernatant solution (b) in the 50 L fedbatch fermentation using the strain G/GMH1
3 讨 论
为了获得具有工业生产价值的GOD高产菌,我们采用黑曲霉的葡萄糖氧化酶蛋白序列,经过密码子优化,构建分泌表达GOD的P. pastoris重组菌G/GOD,并通过提高抗生素浓度,筛选得到高产量重组菌G/GODM,在摇瓶中GOD分泌产量可达5 843.2 U/g,为G/GOD最高产量的8.2倍。在此基础上,进一步构建了多个共表达辅助折叠因子和强化碳代谢途径的第2代高产菌(表2),其中,共表达HAC1的重组菌G/GMH1单位菌体酶活最高,为9 022.1 U/g。在5 L和50 L反应器中,重组菌G/GMH1的胞外GOD产量可达到了7 030.4 U/g和6 656.6 U/g。与已报道GOD表达水平比较,重组菌G/GMH1胞外单位菌体酶活为目前报道摇瓶培养和反应器水平最大值,显示了工业应用的潜力。
围绕提高GOD在P. pastoris的分泌表达,我们尝试了增加基因拷贝数、共表达辅助折叠因子和强化碳代谢途径的策略,其中,增加GOD基因的拷贝数和共表达UPR靶基因的转录因子HAC1取得了最显著的促进作用。这一结果表明,增加基因剂量可有效促进外源蛋白的表达,但过量的外源蛋白表达有可能导致蛋白无法及时进行正确折叠和转运,此时共表达HAC1不仅能够刺激蛋白酶的表达,降解折叠错误的蛋白,而且可以调控UPR响应下游基因的表达,促进蛋白的正确折叠和转运。此外,共表达PDI1和PDI2蛋白也可增强GOD的分泌表达,可能是因为GOD蛋白的186和228位点存在二硫键,这两种二硫键异构酶蛋白有助于二硫键形成,促进了蛋白的正确折叠和分泌。
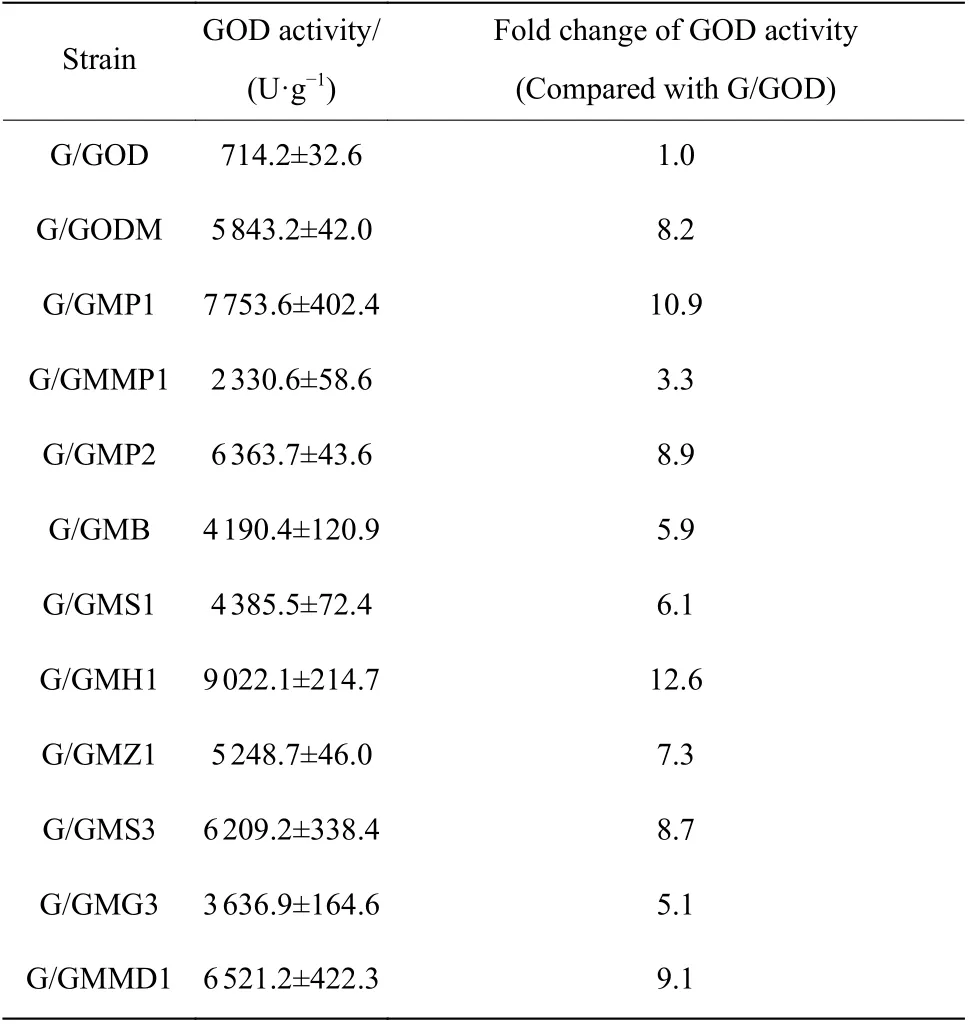
表2 所构建重组菌的胞外单位菌体GOD酶活Table 2 Extracellular GOD specific activity of the recombinant strains
对碳代谢途径进行改造时,强化三羧酸循环循环的苹果酸脱氢酶基因MDH1和磷酸戊糖途径的6-磷酸葡萄糖酸内酯酶基因SOL3均能提高GOD的胞外产量。MDH1催化苹果酸向草酰乙酸的转化,并伴随NAD+向NADH的还原,可增强细胞的能量代谢,为蛋白合成提供ATP[19]。SOL3是磷酸戊糖途径途径中第二步关键酶,强化SOL3的表达有可能提高磷酸戊糖途径途径的通量,为蛋白质合成提供更充足的还原性辅因子NADPH。强化SOL3和MDH1基因有利于外源蛋白的表达,这与Nocon等[15]通过模型预测得到的结果一致,也说明在外源蛋白高效表达时,需要对P. pastoris的胞内碳代谢进行改造以适应蛋白合成的需求并促进其表达。在我们分别强化辅助折叠因子和碳代谢途径取得正效应的基础上,后续可尝试多基因组合的优化策略,或通过蛋白质改造提高GOD的催化活性,来构建下一代的高产菌。
在得到GOD高产菌后,我们进一步在反应器规模验证G/GMH1的性能。虽然共表达HAC1基因后,重组菌的生长稍低于出发菌G/GODM,但其单位菌体产量远高于出发菌,在50 L规模的反应器中最高体积产量达到1 150.5 U/mL。但是,随着发酵规模的扩大,G/GMH1单位菌体酶活出现下降的趋势,在50 L反应器中,GOD的最高胞外单位菌体酶活为6 656.6 U/g,仅为摇瓶最高水平(9 022.1 U/g)的73.4%。这一现象表明,G/GMH1的高产性能并没有在发酵罐中得以实现,重组菌在反应器中的发酵过程还需进行优化。后续可通过优化培养基配方,调整和优化过程控制参数,为重组菌的生长和蛋白表达提供更适宜的环境,充分发挥G/GMH1的高产性能。
