SLC40A1基因突变致遗传性血色病1例报告
2021-05-17朱承谕封忠昕
朱承谕,陈 琦,封忠昕
遵义医科大学附属医院 血液内科,贵州 遵义 56300
遗传性血色病( hereditary hemochromatosis, HH) 是一种罕见的常染色体遗传病,北欧高加索人群发病率较高,其发病机制为铁代谢相关基因突变导致小肠对铁吸收过多,体内过量的铁进行性蓄积造成肝、肾、脑、胰腺等组织损伤,从而引起器官功能障碍导致肝硬化、糖尿病、心肌病、垂体损伤、睾丸萎缩、关节疾病和皮、肤色素沉着等[1]。现将本院收治的1例HH患者的临床资料报道如下。
1 病例资料
患者男性,29岁,因“发现肝功能异常4年,CA19-9升高1个月”于2018年11月30日入本院,患者于2014年体检时发现肝功能异常,转氨酶升高(具体值不详),无乏力、腹胀、目黄等不适,未予重视,2018年10月体检发现转氨酶升高,且伴CA19-9升高,为求进一步诊治就诊于本院。既往史:患者既往体健,父亲因“肝病、糖尿病”于2003年去世,姑姑因“血色病”曾于外院行“放血”治疗于2017年去世,余家庭成员体健,无类似病史。查体:生命体征平稳,神志清楚,慢性肝病面容,全身皮肤无黄染,无瘀点、瘀斑,无蜘蛛痣、肝掌,心肺查体无异常,腹软,无压痛、反跳痛及肌紧张,肝未触及,脾肋下约2横指。血常规:血小板总数 89×109/L;尿常规:尿胆原阴性、尿胆红素阴性;大便常规阴性;肝功能:AST 76 U/L,ALT 114 U/L,GGT 133 U/L,TBil 27.2 μmol/L,直接胆红素5.2 μmol/L,总胆汁酸15.14 μmol/L,Alb 47.4 g/L;铁蛋白>1500 μg/L,铁蛋白(稀释)2689.30 μg/L,不饱和铁结合力3.5 μmol/L,总铁结合力51.1 μmol/L,血清铁47.6 μmol/L,24 h尿铁蛋白14.3 μg/L;AFP 2.18 ng/ml,CA19-9 148.1 U/ml;铜蓝蛋白 26.10 mg/dl;性激素结合球蛋白 89.01 nmol/L,雌二醇202.2 pmol/L,总睾酮>52 nmol/L;甲状腺功能阴性;HBV、HCV、HAV、HEV、HIV+梅毒阴性;抗核抗体+抗核抗体谱、自身免疫性肝炎抗体阴性;病毒四项、凝血功能阴性。上腹部CT平扫+增强:肝脏体积增大,肝脏实质密度增高,CT值110~120 HU(图1);腹部MRI+增强:肝脏弥漫性增大、信号减低,脾脏增大且信号均匀(图2)。HH 1/2/3/4型相关基因检测(测序+MLPA):SLC40A1基因杂合变异(图3)。临床诊断:HH。嘱患者低铁饮食、戒酒,同时予保肝、降酶、去铁、放血等治疗。复查铁代谢:血清铁33.8 μmol/L,不饱和铁结合力12.8 μmol/L,总铁结合力46.6 μmol/L,24 h尿铁蛋白4.6 μg/L,铁蛋白>1500 μg/L,铁蛋白(稀释)2010.00 μg/L;肝功能:ALT 54 U/L,AST 40 U/L,GGT 76 U/L,TBil 23.9 μmol/L,直接胆红素5.9 μmol/L,总胆汁酸2.62 μmol/L。

注:密度弥漫性增高,表现为“白肝”。

注:T2W1上的肝信号减少,在MRI扫描上表现为“黑肝”。
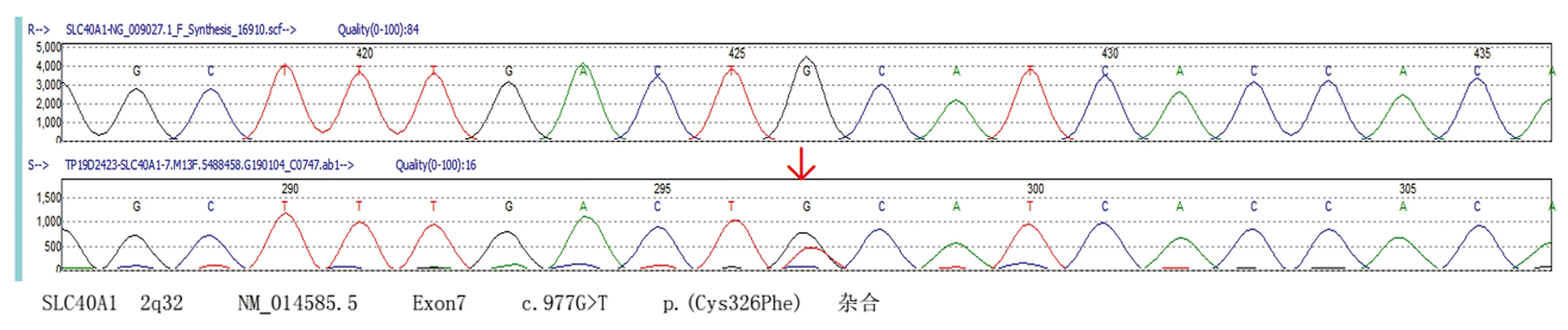
图3 患者SLC40A1基因突变测序结果
2 讨论
本例患者为青年男性,病程较长,无肝硬化、糖尿病、色素沉着等典型表现,既往有明确家族史,查体:慢性病面容,脾脏肋下2横指;辅助检查以肝功能异常升高,铁蛋白明显增高,MRI显示肝脏为低信号,同时排除炎症、代谢综合征、酒精中毒、药物等,基因测序结果提示SLC40A1基因的一个杂合的变异(c.977G>T,p.Cys326Phe),诊断明确后予放血治疗,病情明显好转。
HH表现为肝硬化、糖尿病、皮肤色素沉着、充血性心力衰竭、性腺功能减退等多器官功能障碍,若去铁治疗不及时,最终50%患者将死于肝细胞癌等肝硬化并发症。肝功能受损发生在HH的早期阶段,早期发现并及时予放血、去铁治疗,可预防肝硬化、肝细胞癌的进展,降低病死率。HH的诊断主要基于实验室、影像学检查及HH突变基因检测。在临床中,除了肝功能异常、心功能下降、血糖升高等可能存在的受累器官功能异常外,HH的实验室检查主要反映在铁代谢指标的异常上,估计全身铁存储量最有用的生物标志物是血清铁蛋白,当铁蛋白升高(男性≥300 mg/L,女性≥200 mg/L)时,有必要考虑是否有发生HH的可能性,但不包括炎症、代谢综合征、酒精中毒、肿瘤等。同时,当铁沉积到一定程度时,肝脏CT检测表现为弥漫性的密度增高,类似肝脏增强后的表现,又称“白肝症”,CT值达75~130 Hu。行MRI确定器官中的铁沉积,表现为沉积器官内小颗粒低信号影,在T2加权像降低更为明显,进行肝脏检测时,铁的超顺磁性使肝组织T2驰豫时间缩短,肝脏信号明显减低,似“黑肝症”。肝活检可以进一步明确肝脏病变的程度,随着HH突变基因检测的应用,肝活检的重要性已不及以往,目前主要限于有鉴别诊断需求及肝硬化/进展性肝纤维化高危HH患者肝组织学程度的确认,以帮助评估预后和调整监测/治疗策略。放血疗法是HH的标准治疗方法。血清铁蛋白水平的目标值为50~100 mg/L,根据患者的耐受性、血红蛋白水平和铁蛋白水平调整放血的时间间隔,目前推荐HH患者行1次/1~2周的放血治疗,如果患者不能忍受放血疗法,可以考虑铁螯合疗法。红细胞穿刺术可用于治疗HH,但在临床实践中很少使用[2]。铁调素作为一种小分子多肽类激素,对机体铁代谢起负调控作用,在HH的发生发展中发挥着重要作用[3],对铁调素的不断深入研究发现铁调素替代疗法可进一步从分子水平上改善及治疗HH,人们对开发铁调素疗法的兴趣越来越高[4]。
HH是铁代谢相关的遗传疾病,其遗传基础主要可分为HFE基因突变和非HFE基因突变,可进一步分为4种类型[5]。1型为 HFE 基因突变所致的血色病,在欧美人群中多见,是经典型,该基因纯合或杂合突变中约95%存在纯合C282Y突变;2 型为 HJV 基因和 HAMP 基因突变所致的血色病,其中前者被进一步划分为 2A型,后者被划分为 2B 型;3 型为 TFR2 基因突变所致的血色病;4型为SLC40A1基因突变所致的血色病。4型与前3型遗传特征不同,为常染色体显性遗传,1~3型与铁调素的表达改变或减少有关,而4型则是由膜铁转运蛋白(FPN)突变引起,FPN通过控制肠细胞和巨噬细胞来介导铁的跨膜输出过程,若表达降低会致细胞内游离铁增加,对机体造成影响,Pietrangelo[6]在2017年建议将SLC40A1突变所致的HH分为两类:功能缺失型(4A型)和功能获得型(4B型),前者使FPN不能定位在细胞表面,使铁向外转出受阻,进一步沉积在组织巨噬细胞中,临床上常表现为血清铁、转铁蛋白饱和度降低,MRI易出现“黑脾”,该类型放血治疗疗效不佳,且易并发贫血;后者虽然定位于细胞表面,但不能与铁调素相结合,造成铁调素抵抗,又被称为运铁素血色病,使血液中铁增加并在肝脏沉积。SLC40A1的错义突变最早是由意大利学家于2001年首次报道[7]。一项系统荟萃分析[8]报告称,SLC40A1突变(即V162del、D157N、D181V、G80V、Q182H和R489K)减弱铁转运蛋白的铁输出功能,是4A HH型的常见突变类型。相反,Y64N、V72F、Y501C、N144H突变出现铁调素抵抗,是4B HH型的常见突变,Zhang等[9]发现IVS3+10 del gtt突变是第1个鉴定SLC40A1基因中经典剪接突变的报告,并强调了非HFE突变在亚洲国家HH中的重要性。本文报道的该患者存在SLC40A1基因的一个杂合的变异(c.977G> T,p.Cys326Phe),据报道,C326残基对于铁调素结合必不可少,其破坏性取代导致铁蛋白功能增强,这与发病年龄较早的最严重形式有关。我国目前该基因相关报道较少,有实验[10]显示C326残基突变小鼠表现与HH 4型相似,均出现铁蛋白、转铁蛋白饱和度以及肝脏铁过载表现。Chen等[11]描述了1例69岁的女性患者,具有肝硬化、糖尿病和皮肤色素沉等典型三联征表现,实验室检查提示铁蛋白及饱和度明显上升,基因测序发现相同的功能获得性变体(p.Cys326Phe),家族史中她的两个儿子患有HH,分别在18岁和42岁死于上消化道出血并发症。
综上所述,随着非HFE基因突变HH报道的增加,越来越多的4型患者被诊断出与SLC40A1基因突变有关。对于肝功能长期异常的患者,排除继发性血色病后,结合患者的年龄、家族史、临床表现、伴随症状、疾病的严重程度,还应进一步筛查遗传性肝病。对于可疑患者,进行基因分层诊断。根据测序结果,可以对无症状患者进行早期干预,从而改善患者的预后和生活质量。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:朱承谕负责收集数据、资料分析、撰写论文;封忠昕负责修改论文、拟定写作思路;陈琦指导撰写文章并最后定稿。
