载药脂质体包封率测定方法的研究进展
2021-05-08杭太俊
张 艺,杭太俊,宋 敏
(中国药科大学药物分析系,南京210009)
脂质体是指将药物包封于脂质双分子层中形成的具有纳米结构的新型制剂。包裹药物的脂质双分子层由磷脂和胆固醇组成,其结构内核由磷脂分子的头部聚集而成,具有亲水性,可包裹水溶性药物[1],如已上市的盐酸多柔比星脂质体注射液,其较传统剂型靶向性有所增强,不良反应明显降低[2]。磷脂分子的尾部平行排列组成亲脂性的圆环结构,可携带脂溶性药物。例如,目前已上市的注射用紫杉醇脂质体,它是将脂溶性药物紫杉醇包裹在脂质体中,药物溶解性得以增加,同时避免了聚氧乙烯蓖麻油和无水乙醇混合溶媒的使用,极大地减轻了溶媒所引起的过敏反应[3]。
包封率是脂质体的关键质量属性,它指的是包封在脂质双分子层中的药物含量占总投药量的百分比,能反映出脂质体中药物包封程度的高低,以指导制备工艺的改进。
由于包封的药物性质不同,脂质体膜材料不同,测定每种脂质体包封率的最佳方法往往需要经实验考察才能确定。包封率测定的关键是将包封药物和未包封的游离药物分离,再利用光谱、色谱等分析手段检测包封药物或游离药物的浓度。
常用的包封率测定方法有:离心法、超滤离心法、葡聚糖凝胶柱法、微柱离心法、透析与反透析法、鱼精蛋白凝聚法等[4]。
1 常用的包封率测定方法
1.1 离心法
按照离心转速的不同,离心法分为低速离心法和超速离心法。低速离心法适用于脂溶性药物[4],其工作原理是脂溶性的游离药物不溶于溶解脂质体所需的水相介质,而悬浮在体系中,采用相对较小的离心强度和较短的离心时间,这些未溶的游离药物会因离心力的作用而沉降,但脂质体仍存在于上清液中,从而实现分离。
Sun[5]合成了乙酰半胱氨酸衍生物XXH-32,其难溶于水,传统剂型给药后生物利用度低,于是将其制成脂质体。在测定XXH-32脂质体的包封率时考察了1 000、1 500、2 000、2 500和3 000 r/min 5种离心转速,2 000 r/min时分离度最大,故最终选择了2 000 r/min为离心转速;还考察了2、4、6、8和10 min 5种离心时间,发现离心6~10 min,分离效果差别不大,所以选择离心6 min。实际应用中,上清和沉淀常常不易完全分离,故低速离心法适用范围相对有限。
超速离心法要求离心转速大于20 000 r/min,离心时间一般要长于30 min。离心过程中因空气摩擦产热,需使用控温离心机。与低速离心法刚好相反,超速离心法中脂质体最终存在于沉淀中。由于脂质体和不溶的游离药物会一起沉降下来,无法实现二者的分离,所以此法适用于水溶性较好的药物的测定,可保证游离药物仍存在于上清液中。此外应注意较强的离心力作用可能会导致微粒聚集,破坏脂质体双分子层结构,导致药物发生泄漏[4]。Lopez-Pinto等[6]发现在制备脂质体时可以通过增加胆固醇的用量来提高脂质双分子层的硬度,以减小离心力对脂质体结构的影响。Wang等[7]采用超速离心法测定了盐酸多柔比星脂质体的包封率:取盐酸多柔比星脂质体1 mL,以200 000 r/min转速离心30 min,收集上清液并用生理盐水定容至5 mL,测定其中游离药物质量,再根据含药总量计算出包封率。Han等[8]发现脂质颗粒与柠檬酸钠能够形成絮状物,离心后此种絮状物会上浮,而游离药物仍存在于溶液中,利用此性质可将脂质体和游离药物分离。此外,还考察了加入的柠檬酸钠浓度,发现加入浓度越高,溶液密度越大,脂质颗粒上浮的越多,和游离药物分离就越完全。对于紫杉醇阳离子脂质纳米粒,加入10%的柠檬酸钠分离最优;而对于多柔比星脂质体,加入20%的柠檬酸钠分离最优。
Xu[9]综合了两种离心方法,即先进行低速离心,再超速离心,更为准确地测定了有机金属铱配合物脂质体包封率的数值。先将有机金属铱配合物脂质体混悬液在4℃条件下以5 000 r/min的转速离心10 min,未包封的金属铱配合物被沉淀下来,再将上清液用35 000 r/min的转速离心1 h,以去除上清液中的脂质体部分,再分别测定低速离心后沉淀中和超速离心后上清液中的药物含量,二者总和为脂质体中游离药物的含量。
1.2 超滤离心法
超滤离心法是将脂质体放入配有超滤膜的超滤管中,在适宜的转速下离心,游离药物在离心力的作用下可通过超滤膜,而脂质体则被截留,从而实现二者的分离。
超滤离心法多用于测定水溶性药物脂质体的包封率。而Sui等[10]利用该法成功测定了脂溶性药物甘草次酸脂质体的包封率:取甘草次酸脂质体溶液0.5 mL,以10 000 r/min的转速超滤离心30 min,再检测超滤液中游离甘草次酸的浓度并计算包封率。还分别进行了游离药物回收率以及游离药物和空白脂质体混合物的加样回收率试验,甘草次酸的回收率均在97%以上,说明游离的甘草次酸可以完全通过超滤膜。Mu等[11]制备了吡唑类活性物质XY-4阳离子脂质体,并探究了不同超滤时间和离心转速对包封率测定的影响,最终确定超滤条件为4 000 r/min离心10 min。Gao等[12]制备了长循环吗啡脂质体,采用超滤离心法测定其包封率,离心条件为以10 319 r/min的转速离心5 min,不更换超滤单元,重复超滤离心3次,取最后一次超滤液稀释后进样,测定其中游离吗啡浓度,并计算包封率。
然而“浓差极化”现象的存在限制了超滤法的应用。浓差极化现象是由于在超滤过程中,溶剂和小分子溶质能透过超滤膜,而大分子溶质被截留在膜内,这就会导致超滤膜表面的大分子溶质浓度升高,引起膜附近的渗透压增加,阻碍溶液继续向超滤膜方向扩散,进而降低了溶剂和小分子物质的膜透过率。两性霉素B是一种常用的抗真菌药,已有多家药企研制出两性霉素B脂质体并成功上市。Ran等[13]制备了两性霉素B脂质体,超滤离心后,将获得的超滤液进行HPLC分析,结果未测到两性霉素B。Ran等认为脂溶性较强的两性霉素B在水中形成的大分子聚合物,脂质双分子层和其他有黏性的辅料都可能导致了浓差极化层的形成,从而降低了膜的渗透性,使得游离小分子药物的透过率减小。对样品进行稀释,从而减小磷脂类辅料的黏度可在一定程度上减轻浓差极化现象,但是对于包载脂溶性药物的脂质体,稀释不但会降低游离药物浓度,还可能导致“渗透压休克”[14],即因脂质体膜内外渗透压不一致而导致脂质体泄漏。若稀释后发生了渗透压休克,还可以尝试通过增大离心力来增加游离药物的膜透过率。但传统的超滤膜在高离心力作用下容易发生破损,而中空纤维超滤离心装置凭借其中空结构可以耐受更高的离心力,并且由于中空纤维膜一直浸没在样品溶液和超滤液之间,游离药物可以自由通过超滤膜,因此能够较好地避免浓差极化现象[15]。Xu等[16]利用中空纤维超滤离心法测定了注射用两性霉素B脂质体的包封率,高、中、低三个水平的两性霉素B对照溶液经过中空纤维超滤离心之后,回收率约为98%,空白脂质体加样回收率为100%左右,可见中空纤维超滤离心法更适用于两性霉素B脂质体包封率的测定。
1.3 葡聚糖凝胶柱法
葡聚糖颗粒在溶胀后可形成凝胶状结构,其内部具有一定大小的孔径,小分子游离药物可进入孔内,实现一定程度的保留;而脂质体粒径大于凝胶孔径,脂质体无法进入孔内,于是少量洗脱液便可将其洗脱下来。利用脂质体和游离药物在葡聚糖凝胶柱上保留能力的不同可实现二者的分离。Li等[17]制备了转铁蛋白修饰的长春新碱-粉防己碱脂质体,采用葡聚糖G-50凝胶柱测定其包封率。由于该批脂质体包封率较高,导致含量较低的游离药物在洗脱曲线上未出现明显的洗脱峰,于是决定向脂质体中加入一定量的长春新碱对照品,以增加游离药物含量,使洗脱情况能够更清晰地呈现出来。Li等在0.5 mL脂质体中加入了0.2 mg长春新碱对照品,混匀后加于凝胶柱上,以PBS缓冲液进行洗脱,收集20管洗脱液,测定其中长春新碱的含量并绘制洗脱曲线,从洗脱曲线上可以看出脂质体和游离长春新碱完全分离。由于Sephadex G-50在遇到有机溶剂时易发生脱水,因此Zheng等[18]采用更耐受有机溶剂的Sephadex LH-20凝胶柱分离阿霉素与他莫昔芬双载药脂质体和游离药物,洗脱液选用了脂质体的外水相HEPES缓冲液以及甲醇。Xu等[19]比较了葡聚糖凝胶柱法和中空纤维超滤离心法测定吲哚美辛与维生素A共载脂质体的包封率,发现葡聚糖凝胶柱法测定的包封率值要低于中空纤维超滤离心法;分析原因为葡聚糖凝胶柱法中加入的大量洗脱液稀释了整个脂质体系统,破坏了脂质体和游离药物原有的动态平衡,脂质体发生了泄漏,因而测得的包封率值较低。故在使用此法时,一定要密切关注脂质体结构的完整性。若实验效果不佳,可用微柱离心法替代。
1.4 微柱离心法
相比葡聚糖凝胶柱法,微柱离心法加入的洗脱液体积大大减小,避免了脂质体因稀释作用而发生的泄漏。此法是将溶胀好的葡聚糖凝胶或经预处理的离子交换树脂装入注射器中,反复平衡、离心后制成干燥的微柱,然后在其顶端加入脂质体混悬液,静置几分钟后再加入洗脱液,设置适宜的离心转速将脂质体洗脱下来[20]。若凝胶对脂质体有很强的吸附作用,则不能选用该法。实验中应注意离心转速的选择,转速过大或过小都可能引起凝胶柱柱头断裂,且转速过大会将游离药物也洗脱下来。Song等[21]制备了表面修饰唾液酸的唑来膦酸与多柔比星共载脂质体,采用阳离子交换纤维微柱法测定其包封率,离心条件为以2 000 r/min的转速离心4 min,再用重蒸水洗脱2次,测定洗脱液中包封药物浓度,并计算包封率。Xu等[22]对预饱和Sephadex G-25微柱所需的空白脂质体用量及预饱和次数进行了考察,发现当预饱和空白脂质体用量为150µL时过柱率已达到100%,说明此时微柱对空白脂质体的吸附已饱和,所以最终预饱和用量选定为150µL;预饱和3次时,空白脂质体的过柱率最接近100%,故确定预饱和次数为3次。因此当脂质体浓度较低时应注意对预饱和条件进行考察,以减少柱分析过程中脂质体的损失。
1.5 透析与反透析法
透析法是将脂质体放入截留一定分子量的透析袋内,一般用水或PBS缓冲液作为透析介质,游离药物因透析袋内外的浓度差而向透析介质中转移,而脂质体因为粒径较大则被截留在透析袋内,二者因此实现分离。但脂溶性游离药物在透析介质中溶解度较差,会聚集在透析膜表面,堵塞膜上微孔而无法进入透析外液。
Mura等[23]为增加脂溶性药物苯佐卡因在透析介质中的溶解度,将50%乙醇作为透析介质。Feng[24]将黄芩苷对照品溶液1 mL置于透析袋内,再放在PBS缓冲液250 mL中进行透析,在0~13 h范围内,每小时取透析液3 mL测定其中药物含量,并绘制透析曲线,发现7 h时已透析完全,于是将透析时间设定为7 h。随后进行的回收率试验中,高、中、低3个浓度的回收率均在97%以上,说明此法可用于黄芩苷脂质体包封率的测定。
透析试验中为了满足漏槽条件,需要大量的透析介质,这无疑会稀释整个脂质体系统,破坏脂质体和周围游离药物的动态平衡,甚至导致脂质体的泄漏,并且较长的透析时间也对脂质体的稳定性有很高的要求。反透析法可以避免上述问题的发生。它是将脂质体放在透析袋外,透析袋内装入透析介质。由于透析介质用量大大减少,可有效避免脂质体因为稀释作用而发生的泄漏。Wang等[25]制备了β-紫罗兰酮脂质体,在测定包封率时取20%乙醇5 mL置于透析袋内,再将脂质体混悬液5 mL用20%乙醇水溶液稀释至200 mL,把透析袋放入其中进行透析,12 h后测定透析液中游离β-紫罗兰酮的浓度。Bai[26]比较了透析法和反透析法对马来酸桂哌齐特脂质体包封率的测定效果,发现透析法中游离药物一直难以达到透析平衡,而反透析法中透析150 min后基本达到透析平衡,决定选用反透析法测定包封率。
1.6 其他方法
包封率测定方法还包括:鱼精蛋白凝聚法、固相萃取法、荧光法等。
鱼精蛋白是一种碱性蛋白质,带正电,它可与带负电或中性的脂质体结合形成聚合物,密度有所增加,离心后脂质体-鱼精蛋白聚合物被沉淀下来,因而与游离药物实现分离。Fu等[27]将黄藤素纳米柔性脂质体1.5 mL与鱼精蛋白溶液(10 mg/mL)1.5 mL混合,摇匀后静置3 min,再进行离心,之后将沉淀用曲拉通X-100甲醇溶液2 mL处理后进样测定。可以观察到经过鱼精蛋白沉淀后,原来均一的脂质体混悬液分层明显,上层药液澄清透亮,下层沉淀紧密,便于后续分离操作。Liu等[28]自制了多西他赛脂质体,并比较了高速离心法和鱼精蛋白凝聚法测定结果的差异,同时考察了加入不同浓度的增溶剂吐温80对包封率测定结果的影响;从实验结果可以看出高速离心法测定的结果要低于鱼精蛋白凝聚法;此外随着吐温80浓度增加,两种方法测定的包封率结果均减小。Liu等认为高速离心作用和表面活性剂吐温80,均可能会破坏脂质体原有的亚微米级结构,导致包封率测定值偏低。而鱼精蛋白凝聚法利用的是脂质体的电荷性质,不会对其结构产生影响。
固相萃取法利用的是色谱吸附原理,游离药物被吸附在极性相近的SPE柱固定相上,而脂质体在SPE柱上无保留,少量水便可洗脱下来。Deshpande等[29]利用Oasis HLB固相萃取小柱成功分离了血浆中的两性霉素B脂质体和游离药物,其在待分离血浆混合物中加入了0.1%氨水溶液500µL,作用是增强游离药物在固定相上的保留。但是游离药物在高浓度点的回收率不理想,怀疑是氨水溶液加入量不足,于是在收集到的洗脱液中再加入0.1%氨水溶液250µL,再将这部分洗脱液重新上样,游离的两性霉素B被SPE柱固定相充分吸附,获得了满意的回收率结果。Zhang等[30]发现市售的C18固相萃取小柱常常会吸附紫杉醇脂质体,导致包封率测定结果偏低,而且由于市售固相萃取小柱中的填料孔径小而致密,大粒径的脂质体很难被洗脱下来,于是自制了一种固相萃取柱。为了减弱填料对脂质体的吸附,Zhang等使用了较大粒径的填料(40~60µm),并且减小了装填的紧密程度,此外还对固相萃取柱下端筛板进行了钝化处理,最终获得了满意的结果。固相萃取法对脂质体和游离药物的分离度较高,因为它是借助更特征的,更稳定的吸附能力的差异实现分离。然而该法较为复杂,药物与SPE柱固定相之间吸附作用的强弱受多种因素影响,需要进行大量实验才能摸索出最佳的实验条件。
目前使用的包封率测定方法大部分都需要先将脂质体和游离药物分离,而荧光法不需进行分离,只要利用包封药物和游离药物不同的荧光特性,比较脂质体破乳前后荧光强度的变化,便可计算出包封率。Meng等[31]将RiboGreen荧光染料加入到共载米铂与核酸miR-34a阳离子脂质体溶液中,游离的miR-34a分子会与RiboGreen染料结合产生荧光,利用外标法可以计算出游离miR-34a的浓度,另取一份脂质体破乳后测定miR-34a总浓度,从而计算出包封率。荧光法专属性强,灵敏度高,但能发生特定荧光反应的化合物较少,限制了荧光法在包封率测定中的应用。
上述脂质体包封率测定案例根据主药溶解性和磷脂类辅料组成的不同总结见表1。
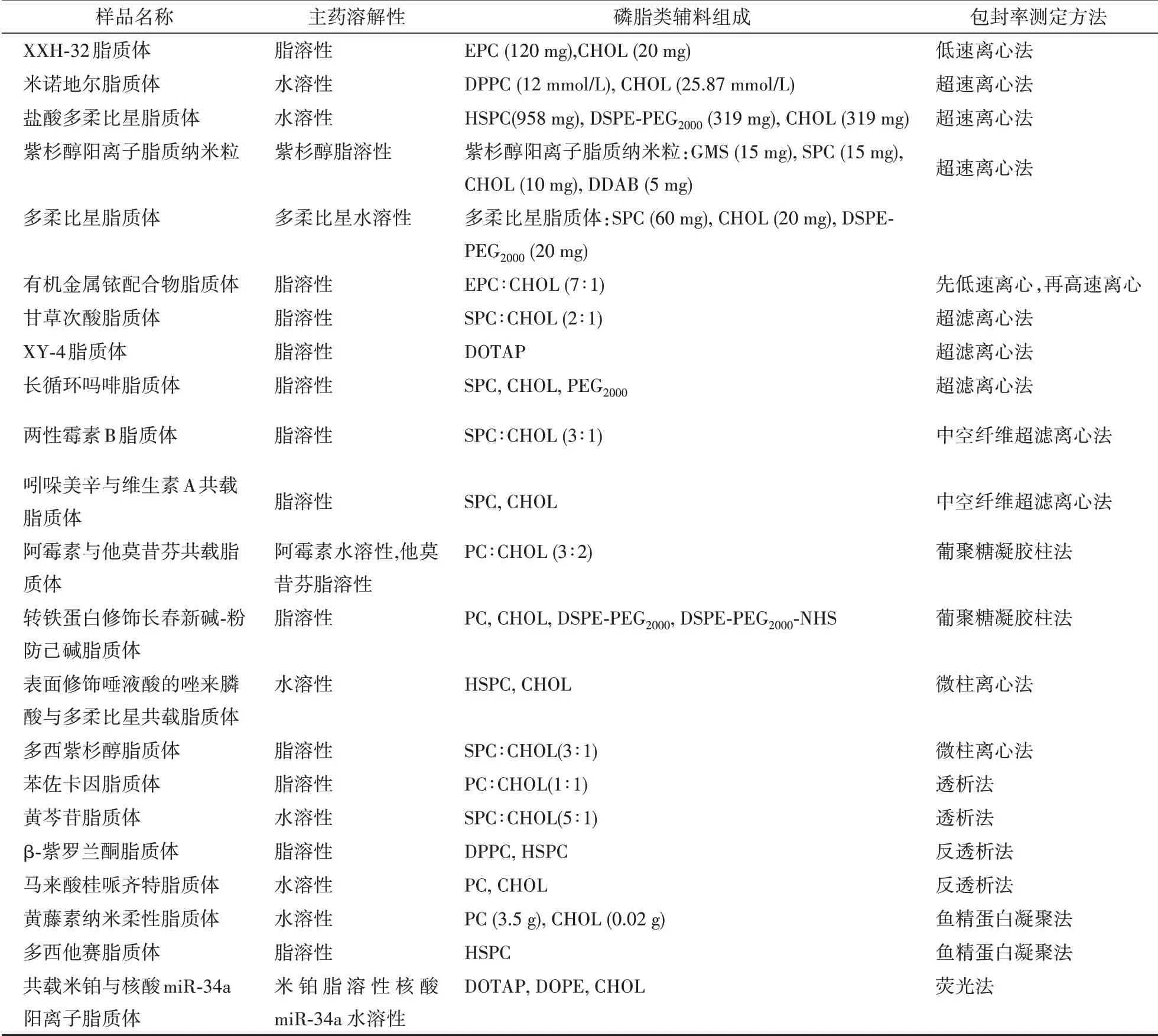
表1 几种载药脂质体包封率测定方法对比
2结 论
本文介绍了多种常见的包封率检测方法,如低速离心法适用于脂溶性药物,操作简单,且不易破坏脂质体膜结构,但脂质体和游离药物常常不能完全分离;超速离心法多用于水溶性药物,且要求脂质体膜结构有一定的硬度,能够耐受高转速的影响。超滤离心法适用范围较广,超滤膜的材料和截留分子量有多种类型,可满足试验者的多重需要;其缺点是可能会出现浓差极化现象,在一定程度上稀释脂质体待测溶液可以避免该现象的发生。葡聚糖凝胶柱法和微柱离心法均是利用分子排阻的原理来分离,但如果药物在凝胶柱上无保留或吸附过强,则不能采用这两种方法。葡聚糖凝胶柱法由于大量洗脱液的加入稀释了脂质体系统,可能会导致脂质体泄漏。但是微柱离心法的柱体积小,加入洗脱液的体积也少,不会引起脂质体泄漏,但是需要考察离心转速,以获得最佳的分离效果。透析法也是测定包封率的常用手段,适用于水溶性药物,但是透析时间一般长达36 h以上,对脂质体稳定性有较高要求;此外大量透析介质的加入同样会稀释脂质体系统,也可能导致脂质体泄漏。反透析法极大地减少了透析介质的用量,较好地避免了因稀释作用而发生的脂质体泄漏。鱼精蛋白凝聚法只适用于带负电及中性的脂质体,而固相萃取法虽然在分离脂质体和游离药物方面有不错的效果,但是实验方法复杂,不易摸索出最佳分离条件。荧光法可在脂质体和游离药物共存的状态下测定包封率,不要求把二者分离,但一般均需要引入荧光指示剂,发生某种荧光反应来计算包封率。
根据待测物的性质和各种测定方法的特点,总结出脂质体包封率测定方法选择决策树,见图1。
3展望
现有的包封率测定方法基本上都是利用脂质体和游离药物在粒径尺寸方面的差异先将二者分离,再准确测定某一方的浓度。但是这种物理差异专属性不强且不稳定,易受影响和干扰。鱼精蛋白凝聚法借助电荷吸附作用增大了脂质体和游离药物的物理差异,使二者分离得更完全;SPE法和离子交换色谱法利用脂质体和游离药物在色谱固定相上保留能力的差异实现了分离,也是一种较好的选择;荧光法不需要分离脂质体和游离药物,通过比较破乳前后荧光强度的变化便可计算出包封率。
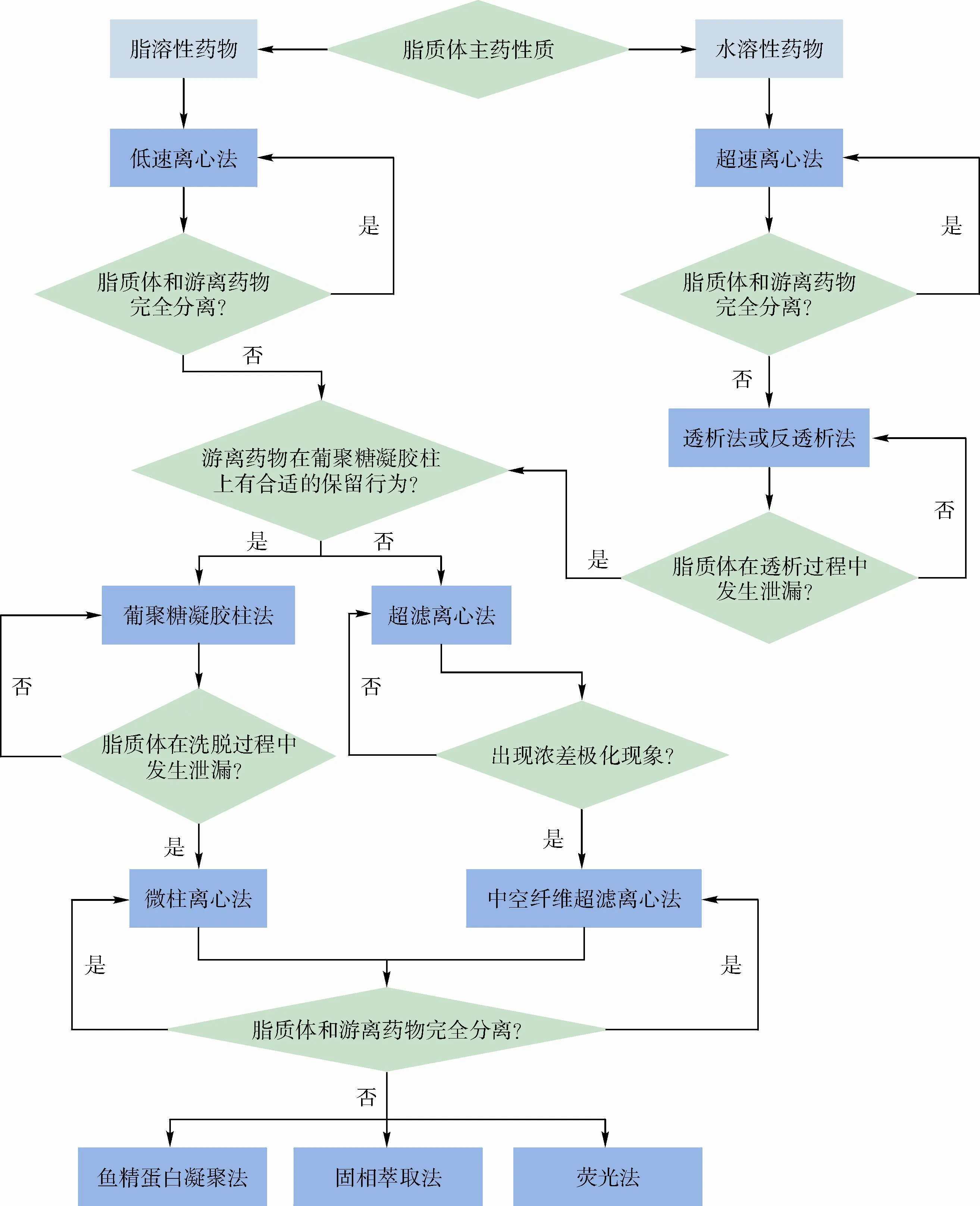
图1 脂质体包封率测定方法选择决策树
未来包封率测定方法应该更多地结合光谱、色谱等手段,以增加方法的专属性和可靠性,还可利用脂质体和游离药物在物理、化学性质方面其他的不同点,开发出具有不同工作原理的新方法,并且各种方法之间可以相互印证,使测定结果更可靠。
