Effect of Outer Carbon Layer Thickness of Carboncovered N-doped Hollow Carbon Nanospheres on Its Electrocatalytic Performance
2021-04-16TAOBowenPANXiangchuanZHANGHaining
TAO Bowen, PAN Xiangchuan, ZHANG Haining
(State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China)
Abstract: Hollow nitrogen-doped porous carbon materials covered with different thicknesses of carbon layers were synthesized to assist evaluation of the influence of nitrogen atom on the surrounding carbon atoms.The designed carbon-based materials were synthesized through pyrolysis of surface-attached block copolymer layers on silica nanoparticles with different thicknesses of the second block of grafted polymer chains, followed by removal of silica templates. The experimental results reveal that coverage a carbon layer with proper thickness can improve the oxygen reaction reduction activity of nitrogen-doped carbon materials as evidenced by the positive shift of half-wave potential in linear scanning voltammetry response curves. The conclusions may provide a reference work on understanding the active sites and designing materials with superior electrochemical performance.
Key words: surface-attachment; porous carbon; block copolymers; surface coverage; oxygen reduction reaction
1 Introduction
With the rapid commercial development, there appears large amount of environmental problems such as global warming, photochemical smog, and acid rain,which have threatened the health and life of human beings. In addition, due to the over-exploitation and ineffective utilization of fossil fuels, the energy crisis is also an impending issue waiting to be solved[1,2].Thus, it is urgent to develop clean and effective alternatives to settle troubles induced by excessive usage of fossil energy. Indeed, many efforts have been put on oxygen-involved electrochemical energy conversion and storage devices including fuel cells[3,4]and metal-air batteries[5,6]due to replace traditional internal combustion engines. However, the sluggish kinetics of oxygen-involved reactions, particularly oxygen reduction reaction (ORR), has hindered the progress of those admirable devices[7,8]. At present, numerous works have been done to develop efficient and cost-effective catalysts for ORR including Pt-based catalysts, heteroatoms doped carbon materials, and transition metals oxides.For platinum-based catalysts, the commercial application is limited by the costly price and scarce resources,in spite of the preferable adsorption and reduction of oxygen during the catalytic process of ORR[9,10]. For transition metals oxides, the improved catalytic activity may derive from the formation of M-N-C/M-C bonds serving as active sites during thermal treatment and it may also ascribe to the promotion of shaping diverse nanostructured carbon[11,12].
However, it is confirmed that carbon materials doped by heteroatoms can acquire reasonable electrocatalytic activity, attributed to the large surface area,easy formation of porous structure or large number of the generated active sites[13,14]. For instance, Chenet alsynthesized porous nitrogen doped graphene layers(NGS) by pyrolyzing chitosan film deposited on the graphitic carbon nitride (g-C3N4) nanosheets, and the as-prepared NGS owned competitive ORR catalytic performance to benchmark catalyst Pt/C. The remarkable behavior might ascribe to porosity of the material,doping, and large surface area with specific value at 1 118.3 m2·g-1for NGS-1000[15]. In addition, Guet alprepared N-doped carbon materials (NCs) by carbonizing citric acid and dicyandiamide. The results suggested that the pore volumes of the NCs enhanced with the increasing nitrogen content of precursor and the halfwave potential of the NC-6 owning superior ORR behavior was just 66 mV negative than Pt/C catalyst[16].
Besides, plentiful research has been performed to understand the active sites of nitrogen doped carbon materials. It has been suggested that it is the adjacent carbon atoms rather than nitrogen atoms themselves that act as catalytic active sites. Due to the discrepancy of electronegativity, the electron density of carbon atoms is redistributed and charges of carbon atoms are transformed accordingly by the introduction of nitrogen atoms. The most controversial focus on catalytic mechanism for oxygen reduction reaction is whether graphitic N[17-19]or pyridinic N[20-22]dominates the catalytic process. For example, Yanget alfound that graphitic N was responsible for oxygen reduction reaction under alkaline condition through Mott-Schottky experiments, ultraviolet photoelectron spectroscopy(UPS) analysis, and spectra X-ray absorption near-edge structure (XANES) spectroscopic measurements. Integrating the work function from UPS and peak change of graphitic N and pyridinic N, it was suggested that N-GRW sample possessing more graphitic N edge sites presented optimum activity for ORR and the improved performance ascribed to the enhancement of oxygen adsorption from higher nucleophilic reaction of carbon atoms induced by graphitic N[23]. However, Guoet alused CO2temperature programmed desorption (TPD)measurements to test the model catalysts including pyridinic N-dominated HOPG (pyri-HOPG) and graphitic N-dominated HOPG (grap-HOPG) at room temperature, proving that it was carbon atoms adjacent to pyridinic N acting as active sites with Lewis basicity that adsorbed O2molecules for ORR in acid electrolyte[24].
Though there have existed large amount of work concentrating on the effect of nitrogen dopant and bonding states of nitrogen atoms on the electrochemical performance, less attention has been paid to the quantitative impact of nitrogen atoms on adjacent carbon atoms. In our previous work, we reported that coverage a thin layer of graphite carbon can improve the ORR activity of nitrogen-doped carbon materials under both alkaline and acidic conditions[25]. To make clear the possible extended depth of nitrogen atoms on carbon atoms, we synthesized hollow porous nitrogen doped carbon materials covered by a thin carbon layer (C@N-C) with different thickness using surface-attached block co-polymer layers as precursors. The ORR activity of samples was then evaluated in detail with respect to the outer carbon layer thickness.
2 Experimental
2.1 Materials and methods
Nano-silica particles (99.5%, average diameter=30 nm,MW=60.08) were received from Aladdin(Shanghai, China) and activated with piranha solution(98wt% H2SO4:30wt% H2O2=7:3 in volume) to increase the amount of hydroxyl groups. Cuprous chloride, methanol, toluene and dichloromethane are all analytical grade and were purchased from Sinoreagent(Shanghai, China). The commercial cuprous chloride was reacted with acetic acid under vigorous stirring continuously for 12 h and then washed with ethanol to remove the cupper chloride, followed by drying in vacuum at 40 ℃ prior to use. Toluene and dichloromethane were distilled over sodium and calcium hydroxide separately by refluxing to remove water and then stored with zeolite. 2-(Dimethylamino)ethyl methacrylate(DMAEMA, 99%, Macklin) was purified through alkaline aluminum oxide column to remove 4-Methoxyphenol before use. Deionized water was obtained using a Ulupure-H ultrapure water generator (Ulup, China)with resistivity of 18.25 MΩ·cm-1at 25 ℃. All the other chemicals including Tris[2-(dimethylamino)ethyl]amine (Me6TREN, 98+%, Alfa Aesar), dimethychlorosilane (96%, Alfa Aesar), 2-bromoisobutyryl bromide(BiBB, 98%, Aladdin), hydrofluoric acid, triethylamine(TEA), N, N-Dimethylformamide (DMF) and ally alcohol (AR, Sinoreagent) were used without further purification .
2.2 Synthesis of initiator modified nano silica (SiO2-BiBPDMCS)
Prior to the copolymerization, the functionalized initiator (3-(2-bromoisobutyryl)propyl)dimethylchlorosilane (BiBPDMCS) for surface-initiated atom transfer radical polymerization (SI-ATRP) were synthesized according to the Ref.[26]. The synthesized initiator was dissolved in anhydrous toluene by stirring under ice bath overnight to obtain homogeneous solution with concentration of 50 mmol·g-1. A desired amount of silica was dispersed in BiBPDMCS/toluene solution for 48 hours using triethylamine (TEA) as catalyst. The modified nano silica was collected after extensively washed with toluene and ethanol and finally dried under vacuum at 40 ℃.
2.3 Synthesis of surface-attached block copolymers (SiO2-PDMAEMA-b-PS)
The surface-attached block copolymer brushes were synthesized by a two-step SI-ATRP approach in which the nitrogen-contained polymer chains was first grown from the surface and pure hydrocarbon polymers was the second block. Typically, 0.5 g initiator modified nano silica was added into a flask containing 10 mL monomer DMAEMA and 6 mL methanol. The mixture was degassed by three cycles of freeze-vacuum-thaw cycles to remove the dissolved oxygen and the system was replaced with nitrogen after the third vacuum pumping. A solution including 30 mg CuCl,200 μL Me6TREN, and 2 mL methanol prepared in glovebox beforehand was then added into the above mixture under protection of nitrogen. After 2 h polymerization at 70 ℃, the reaction was terminated by exposing the reaction system in air. The as-obtained product was washed by ethanol for three times and then with DMF for one time. In the second step, the growth of second block was similar to the first step by replacing the monomer DMAEMA with styrene and raising the reaction temperature to 90 ℃ with different polymerization times. For comparison, the copolymerization was also performed using polystyrene (PS) as the first block and PDMAEMA as the second block of polymer chains.
2.4 Fabrication of C@N-C
The copolymer brushes obtained under different reaction times of the second block were pyrolyzed at 1 000 ℃ under argon atmosphere for 1 hour with a ramping rate of 5 ℃·min-1. The carbonized samples were then immersed in hydrofluoric acid for 48 hours to remove silica templates. The target products were obtained after extensively washing with deionized water and dried under vacuum at 80 ℃.
2.5 Characterization
Fourier transform infrared (FTIR) spectra were recorded on 60SXB spectrometer (Nicolet) with a resolution of 4 cm-1to determine the chemical composition of samples and test the anchoring of block polymers.Raman spectroscopy (INVIA, Renishaw) and X-ray diffraction (XRD, D8 Adwance, Bruker Corp.) were applied to characterize the crystal structure of materials. Elemental analysis was carried out on a Vario EL Cube analyzer (Elementar, Germany) to obtain the nitrogen content of samples. Transmission electron microscopy (TEM) images obtained from H-600 STEM/JEM2100F (Hitachi) were applied to characterize the porous architecture of samples and the thickness of carbon layers. Brunauer-Emmett-Teller (BET) surface area and pore size distribution were determined through nitrogen adsorption-desorption isotherms performed on Micromeritics ASAP 2020 instrument at 77 K. Prior to the measurements, samples were disposed under vacuum at 80 ℃ for 24 h to removed adsorbed gas and free water. X-ray photoelectron spectroscopy (XPS, VG Multilab2000X) was applied to determine the near-surface elemental composition and valence states of elements using Al Kα irradiation source.
2.6 Electrochemical measurement
The electrochemical measurements of samples were conducted on electrochemical workstation (CHI 604D Shanghai, China) equipped with an rotating disk electrode (RDE). Before the electrochemical tests, 5 mg catalyst prepared was dispersed in a solution containing 800 μL isopropanol, 200 μL deionized water, and 40 μL Nafion solution (5wt% in isopropanol, Dupont),followed by sonication for at least 20 min to form a homogeneous solution. Then, a standard three-electrode system using the glassy carbon electrode (GC electrode,5 mm in diameter, Tianjin Aida) coated catalyst ink (20 μL) as working electrode, graphite rod as counter electrode, and Hg/HgO (1 M KOH) as reference electrode was applied to implement cyclic voltammetry (CV) and linear sweep voltammetry (LSV) measurements in 0.1 M KOH saturated with high-purity N2/O2at ambient temperature. The catalyst loading was calculated to be 0.49 mg·cm-2as the surface area of the glassy carbon electrode is 0.196 cm-1.
3 Results and discussion
The designed carbon-based materials were fabricated by pyrolysis of pre-synthesized surface-attached block copolymer brushes, followed by removal of silica templates, as schematically illustrated in Fig.1. As precursors, copolymer brushes were synthesized via surface-initiated atom transfer radical polymerization (SIATRP) technique in which initiator monolayers were first self-assembled onto silica templates. After growth the first polymer block, the second block of polymer chains can be subsequently grafted on the remaining active end of polymer chains since ATRP is a living polymerization process. The outer layer thickness of final carbon-based materials was varied by controlling the layer thickness of the second block of polymer chains in precursor through the change in polymerization time for the second chain growth. To differentiate samples synthesized clearly, the materials were labeled as C@NC-6, C@NC-12, and C@NC-24 corresponding to samples using styrene as the second monomer with reaction time for 6, 12, and 24 h, respectively. The controlling sample using PS as first block polymerized for 2 h and PDMAEMA as second block polymerized for 12 h was denoted as NC@C.
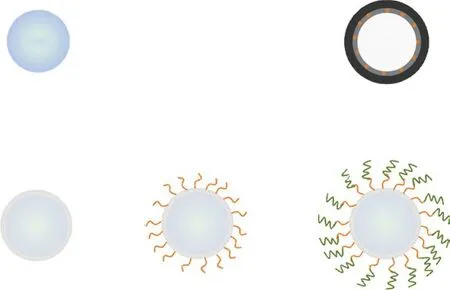
Fig.1 Schematic illustration of synthesis of C@NC. The chemical structure of functionalized initiator BiBPDMCS was displayed in the figure
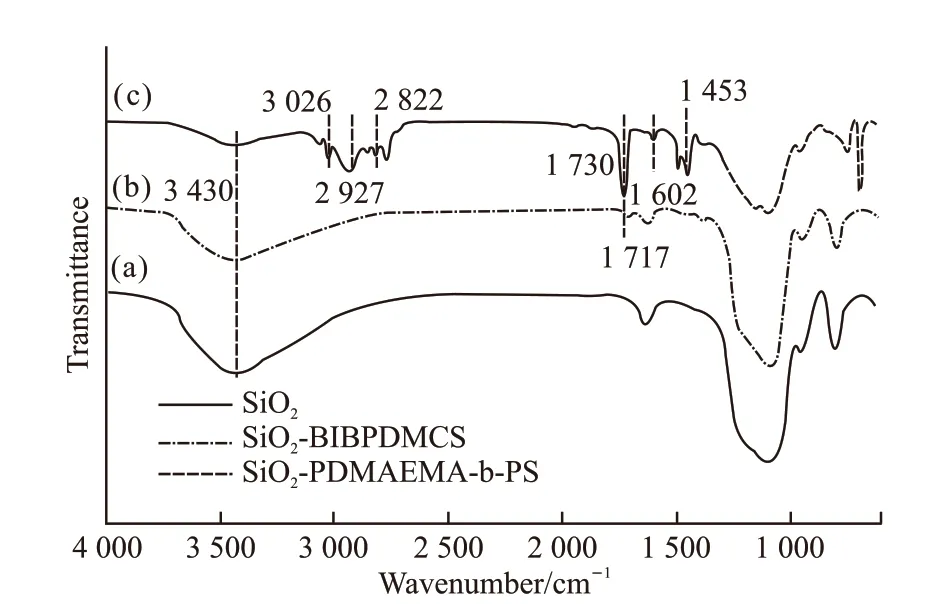
Fig.2 FTIR spectra of bare silica(a), SiO2-BiBPDMCS(b), and SiO2-g-PDMAEMA-b-PS(c)
FTIR spectroscopy was applied to qualitatively determine the synthesis of block copolymer brushes.Fig.2 shows FTIR spectra of bare silica, SiO2-BiBPDMCS, and SiO2-g-PDMAEMA-b-PS. Compared with bare silica, the newly appeared absorption band at 1 717 cm-1corresponded to the stretching vibration of C=O,indicating the successful self-assembly of functionalized initiator[27,28]. For the spectrum of SiO2-g-PDMAEMA-b-PS, absorption bands appeared at 2 927 cm-1,2 822 cm-1were the stretching vibration of saturated C-H. The strong adsorption band at 1 730 cm-1representing the existence of C=O and absorption bands appeared at 1 602 cm-1and 1 453 cm-1are characteristic adsorption peaks of benzene ring, confirming the successful grafting of PDMAEMA and PS blocks[29-31].
The designed carbon-based materials were fabricated by pyrolysis of block copolymer brushes,followed by removal of silica templates. Raman spectroscopy was applied to characterize the carbonization degree of samples received, as shown in Fig.3(a). The observed characteristic absorption peaks at about 1330 and 1 590 cm-1represented the D band and G band,attributing to the disoriented or defected carbon and graphitic carbon, respectively[32,33]. In general, the intensity ratio ofID/IGwas applied to describe the degree of carbonization and higher specific value indicated more disordering structures. The calculatedID/IGvalues for NC@C, C@NC-6, and C@NC-12 were very similar (about 1.06), less than that for C@NC-24 (1.13),suggesting the relatively high degree of disordering for sample C@NC-24. In addition, the diffraction peaks in XRD patterns (Fig.3(b)) appeared at appropriately 24oand 44oattributed to the (002) and (101) planes of partially graphitized carbon[34,35].
XPS surveys are recorded in Fig.4(a) to determine the elemental composition near the surface. It is evident that C1s, N1s, and O1s peaks are observed at 284, 400,and 532 eV for all the tested samples. Moreover, the nitrogen content for C@NC samples decreased from 3.32at% to 0.85at% with the increase in polymerization time of the second blocks from 6 h (C@NC-6) to 24 h(C@NC-24), indicating the increased outer layer thickness of coated carbon layer. To further understand the correlation between the outer layer thickness and bonding state of nitrogen atoms, deconvolution of high-resolution N1s peaks was performed, as shown in Fig.4(b).It can be seen that all the N1s spectra for four samples were deconvoluted into more precise peaks at about 398.5 eV, 401 eV, and 402 eV ascribing to pyridinic N,graphitic N, oxidized N, respectively[36,37]. It was surprising that nitrogen atoms in C@NC-24 consisted of just graphitic N, suggesting that the active sites might be reduced as the carbon layer thickness exceeded a certain value.
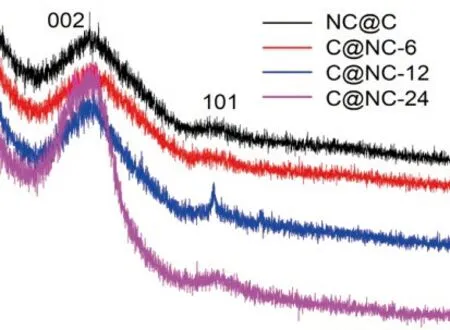
Fig.3 Raman spectra(a) and XRD patterns(b) for the synthesized samples as indicated in the figure
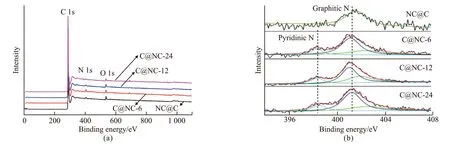
Fig.4 XPS surveys(a) and deconvolution results for high-resolution N1s peaks(b) of NC@C and C@N-C for different polymerization time of second blocks as indicated in the figure
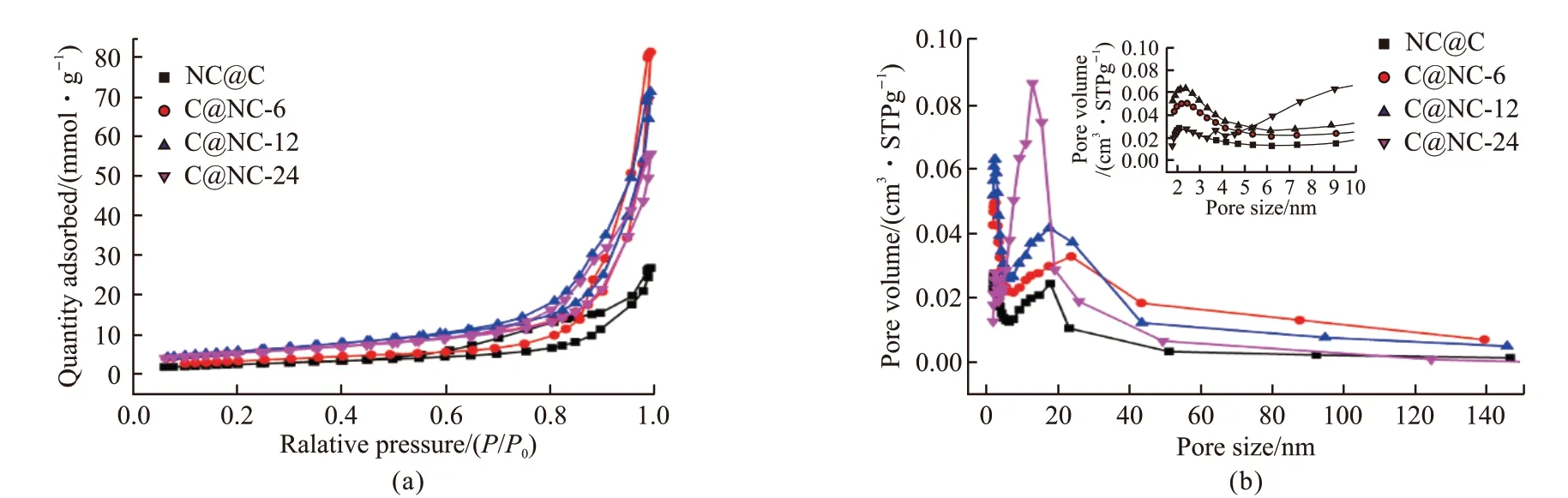
Fig.5 Nitrogen adsorption-desorption isotherms(a) and the derived pore size distribution(b) of NC@C and C@NC with different polymerization of the second block as indicated in the figure
Since porous structure is critical for catalytic activity of materials, N2adsorption-desorption isotherms were recorded to determine the BET surface area and porous parameters of the synthesized samples as displayed in Fig.5(a). It is apparent that all the samples exhibited the characteristic Ⅳ isotherms with type H1 hysteresis loop, indicating the existence of mesopores[38,39]. The accordingly derived pore size distribution (Fig.5(b)) further confirmed the existence of mesopores centered at about 20 nm. In addition, micropores with pore size about 2.5 nm were observed for all the samples (inset in Fig.5(b)), caused by the decomposition of nitrogen-containing polymers as observed in the Ref.[40]. The derived BET surface area values for NC@C, C@NC-6, C@NC-12, and C@NC-24 are 211, 453, 493, and 466 m2·g-1, respectively. Compared to the sample with pure carbon as inside layer,samples with the nitrogen-doped carbon as inner layer exhibited higher surface area because the emission of nitrogen-contained species through the outer layer can lead to the generation of more micropores. Accordingly, samples with nitrogen-doped carbon as inner cores have the large surface aera.
SEM images of C@NC-6 (Fig.6(a)), C@NC-12 (Fig.6(b)), and C@NC-24 (Fig.6(c)) reveal that the formed carbon materials are spherical shapes and aggregation of spherical nanoparticles existed. Apart from the aggregated particles, isolated small particles can be observed with diameters about 25-30 nm (particularly clear in Fig.6(c)), which are consistent with the applied silica templates. Furthermore, TEM images for all the tested three samples (Figs.6(d)-6(f)) show the hollow structure of the synthesized carbon-based materials and the pore diameter is about 20 nm, which agrees with the result of N2adsorption-desorption measurement.The pore diameter is smaller than the diameter of silica templates (average diameter of about 30 nm), suggesting that the formed carbon materials may shrink during the removal of silica templates. Moreover, the stability of samples seems to be improved with the increase in polymerization time of the second block as the spherical particles becomes complete. This was possibly due to the increased thickness of the second block of attached polymer chains and the accordingly formed carbon layers.
To evaluate the influence of carbon layer thickness on the catalytic performance of oxygen reduction,LSV measurements were performed. Fig.7(a) shows LSV curves in oxygen-saturated alkaline solution for C@NC with different polymerization time of the second block. For comparison, LSV curve of NC@C is plotted in the same figure. It is apparent that all the tested samples have very similar onset potential at about 0.838 VvsRHE, indicating the same active sites for all the tested samples including the type of atoms and the bonding state of the according atoms. However, the samples displayed different half-wave potentials, suggesting the different numbers of active sites involved in oxygen reduction reactions. Specifically, the halfwave potential of C@NC-12 is about 12 mV and 19 mV more positive than those of C@NC-6 and C@NC-24, respectively. It has been reported that carbon atoms within a certain distance of doped nitrogen atoms are active sites for ORR and coverage of thin carbon layer can improve the ORR activity[25]. If the outer carbon layer is too thin, the surface density of active sites is possibly low whereas the ORR activity could also be reduced due to the reduced electron density of carbon atoms if the outer carbon layer is too thick. It is therefore understandable that there existed an optimum carbon layer thickness for oxygen reduction reaction catalyst with polymerization time of second chains at 12 h.In addition, LSV curves of C@NC-12 under different rotation rates (Fig.7(b)) and the accordingly derived Koutecky-Levich (K-L) plots (Fig.7(c)) are displayed to elucidate the electrode reaction mechanism. It is evident that the electron transfer number was calculated to be appropriate 4 according to K-L equations, suggesting the four-electron transfer dominated reaction for ORR using the synthesized catalyst. It has been widely accepted that there exited two-electron pathway and four-electron pathway for ORR[41]. For the former one,O2was reduced into H2O2firstly and then transferred to H2O, while the latter one reducing O2into H2O directly.The generation of H2O2is harmful for fuel cell applications since H2O2can accelerate the degradation of membrane and polymeric ionomers. The four-electron transfer dominated ORR for the synthesized C@NC-12 indicates that the generation of H2O2is unfavorable and application of C@NC-12 could be possibly improve the durability of thus-assembled devices.
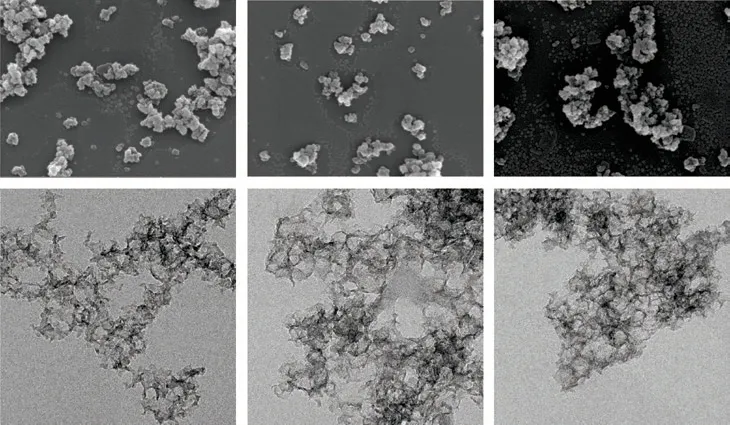
Fig.6 SEM images (a, b, c) and TEM images (d, e, f) of C@NC-6 (a, d), C@NC-12 (b, e), and C@NC-24 (c, f). Scale bars in SEM and TEM images are 500 and 50 nm, respectively
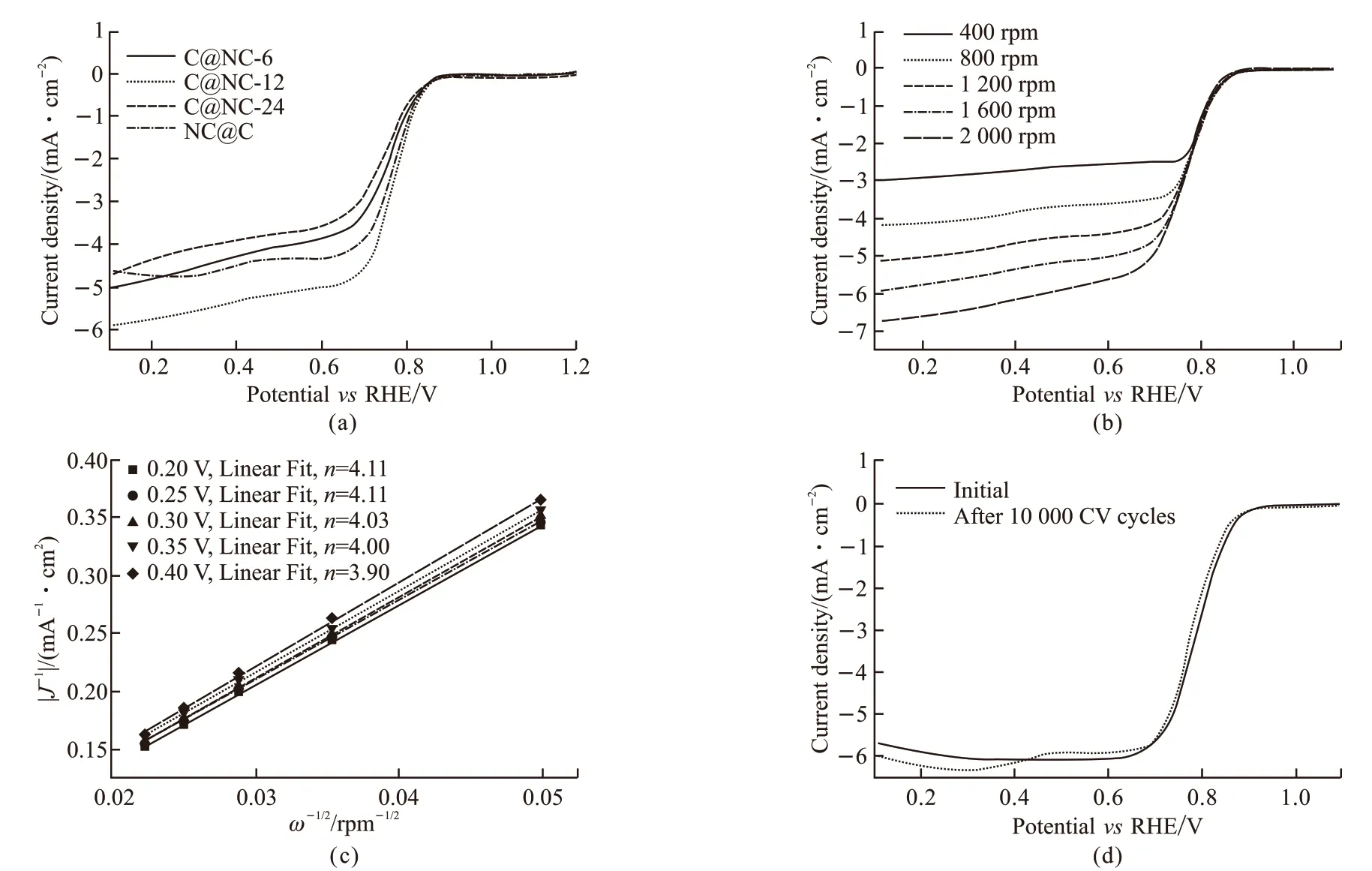
Fig.7 (a) LSV curves for different samples at 1 600 rpm as indicated in the figure; (b) LSV curves and (c) the derived K-L plots of C@NC-12 plots under different rotation speed; (d) LSV curves of C@NC-12 before and after 10000 continuous CV cycles
The stability under electrochemical environment is of great concern for practical applications of electrocatalysts. The stability of the synthesized C@NC-12 was evaluated by continuous cyclic voltammetry(CV) cycles. Fig.7(d) shows LSV curves of C@NC-12 before and after 10000 continuous CV cycles in 0.1 mol·L-1KOH aqueous solution. It can be clearly seen that the difference in half-wave potential values for C@NC-12 before and after 10 000 continuous CV cycles is only about 2-3 mV, even within the experimental error.This result demonstrates that the synthesized C@CN-12 has an excellent electrochemical stability for ORR.
4 Conclusions
In summary, hollow porous nitrogen doped carbon materials covered with virous carbon layer thickness were prepared successfully using nitrogen-contained polymer and polystyrene as nitrogen source and carbon source, respectively. To acquire independent carbon layers, surface-initiated atom transfer radical polymerization was selected for growth of surface-attached block copolymer layers. The designed carbon-based materials were finally fabricated by pyrolysis of surface-attached block copolymer layers on silica nanoparticles, followed by removal of silica templates.The results revealed that carbon atoms within certain distance following zigzag shape from the doped nitrogen atoms are active sites and the surface density of active sites is the key parameter for oxygen reduction reactions as evidenced by the superior electrocatalytic activity of thin carbon layer covered nitrogen-doped hollow carbon nanospheres compared to the sample of nitrogen-doped carbon covered hollow carbo nanospheres. Moreover, it was also found that there existed an intermediate thickness of the covered carbon layer with an outstanding electrocatalytic performance for oxygen reduction since the very thin outer carbon layer leads to low surface density of active sites and the very thick outer carbon layer can reduce the electron density and activity of active sites.
杂志排行

Journal of Wuhan University of Technology(Materials Science Edition)的其它文章
- Comparative Case Study on Adhesion of Three Common Sizing Agents to Cotton and Polyester Yarns
- Ceramification of Composites of MgO-Al2O3-SiO2/Boron Phenolic Resin with Different Calcine Time
- Natural Fresh Proteins Directed Hierarchically Porous Nitrogen-doped TiO2 as with High Performance as Photocatalyts and Electrode Materials
- Dynamic Adsorption of Toluene on Hierarchical Porous Carbons with Varying Pore Structure
- Self-propagating High-temperature Synthesis of Sm and Zr Co-doped Gd2Ti2O7 Pyrochlore Ceramics as Nuclear Waste Forms
- Solidification Behavior of in situ TiB2/Cu Composite Powders during Reactive Gas Atomization