酪氨酸羟化酶缺乏症导致多巴反应性肌张力不全患儿临床及遗传特点
2021-04-14朱海霞李小晶梁惠慈吴汶霖陈连凤陈文雄
朱海霞,李小晶,梁惠慈,吴汶霖,侯 池,陈连凤,田 杨,陈文雄
(广州市妇女儿童医疗中心神经内科,广东广州 520120)
多巴反应性肌张力不全(dopa-responsive dystonia,DRD)是一种罕见的常染色体遗传运动障碍,最初被Segawa 报道[1]。其典型的表现为儿童或青少年发病,症状可有昼夜波动,低剂量左旋多巴治疗可产生戏剧性和持续性的效果。该病的患病率为0.5~1/百万[2]。DRD的主要原因是神经递质多巴胺的缺乏。DRD 涉及的基因包括GCH1、TH、PTS、SPR、QDPR和PCBD[3]。其中,TH基因的缺陷导致酪氨酸羟化酶缺乏症(tyrosine hydroxylase deficiency,THD),在这种情况下,酪氨酸转化为多巴胺的过程被阻断,从而导致多巴胺的生成减少,患儿出现DRD 的临床症状。大多数THD 采用左旋多巴治疗,效果明显,但由于THD 发病率低,临床医生对其认识程度不足,容易误诊。因此,我们总结4 个家系,6 例THD 患儿的临床特征、治疗方案、基因检测结果和随访,以探讨THD 患儿临床表型与基因突变的相关性,提高临床医生对THD 的认识,以促进早期干预,改善患儿预后。
1 材料与方法
1.1 一般资料收集
收集2017年6月至2020年11月在广州市妇女儿童医疗中心小儿神经内科首次确诊的6 例THD患儿。本研究纳入4 个非亲缘的中国血统家系,共6 例患儿。患儿诊断符合THD 的诊断标准或遗传标准[4]。临床标准是指左旋多巴治疗后肌张力障碍改善至少50%的患儿。遗传标准是指基因分析显示TH基因突变的患儿。
1.2 临床资料收集
收集患儿临床资料,包括家族史、性别、发病年龄、诊断年龄、肌张力障碍的临床表现、发病时附加特征、昼夜波动、神经体征、实验室检查(肝肾功能、铜蓝蛋白、血乳酸、血氨、血浆氨基酸、酰基肉碱、尿有机酸、甲状腺功能)、头颅磁共振成像及神经生理测试(脑电图,脑干听觉和视觉诱发电位)、治疗及预后。末次随访时间为2020年11月。所有患儿都接受了一系列神经学检查和发育里程碑评估。末次随访,采用韦氏儿童智力量表,通过智力商数测验,对患儿认知功能进行评分。
1.3 基因分析
本研究经医院伦理委员会批准(穗妇儿科伦批字[2019]第40101 号),并经监护人知情同意。取患儿及其父母外周血各4 mL。以盐析法获得基因组DNA,采用全外显子二代测序方法进行遗传学分析,使用Illumina Nextseq 500 测序平台进行测序,测序平均覆盖深度为200×左右,>10×覆盖区间占100%,>20×覆盖区间占100%,并对检测结果进行Sanger 一代验证。应用预测软件对变异的保守性、致病性、危害性进行预测。参考美国医学遗传学和基因组学学会变异分类指南,对变异进行分类。检索HGMD、PubMed、Clinvar 等数据库,检索相关文献,分析文献内容。
1.4 统计分析
数据输入SPSS 19.0 统计学软件,建立数据库,进行统计学描述。计数资料以例数表示,计量资料以中位数(范围)表示。
2 结果
2.1 6例THD患儿的临床表现
本研究6 例THD 患儿发病的中位数年龄为11.5 个月(范围从3 个月到4 岁)。6 例患儿,男4例,女2 例,男女患儿比例为2:1。6 例患儿均否认家族遗传病史。6 例患儿均存在肌力下降及肌张力异常,运动发育迟缓,2 例面具脸,3 例流涎,2 例晨轻暮重表现,其中患儿#3(弟弟)及#4(姐姐)来自同一家庭,既往外院诊断为“遗传性痉挛性截瘫”,#3 患儿1 岁会爬,2 岁独走,5 岁能跑,但运动耐力差。首次来院时9 岁,行走速度慢且不稳,行走时左上肢内收内旋姿势,吞咽慢,言语及智力发育正常。#4 患儿4 岁出现肢体乏力,5 岁出现易跌倒,9岁需坐轮椅,13 岁完全卧床,有严重的脊柱侧弯,生活不能自理。另外2 例患儿(#5,#6)为其母首次自然受孕,异卵双生的男性患儿,兄弟二人既往被诊断“脑性瘫痪”,长期康复治疗,病情无改善。平素发热后精神运动发育会倒退(表1)。
2.2 辅助检查
6 例患儿肝肾功能、铜蓝蛋白、血乳酸、血氨、血浆氨基酸、酰基肉碱、尿有机酸、甲状腺功能、头颅磁共振成像及神经生理测试(脑电图,脑干听觉和视觉诱发电位)等检查均未见异常。
2.3 6例THD患儿的基因检测结果
对4 个家系6 例THD 患儿行全外显子基因检测,其中患儿#3,#4来自同一家庭,#3为弟弟,#4为姐姐,姐弟两人的TH基因突变位点一致;患儿#5,#6 来自同一家庭,为其母异卵双生,自然受孕的兄弟,兄弟两人的TH基因突变位点一致。在我们的病例系列中发现了TH基因的6 个错义突变以及1个移码突变。6 例患儿的TH基因均为复合杂合突变,与常染色体隐性遗传一致。其中既往文献报道过的突变位点5 个,包括:c.698G>A(p.R233H)、c.1145T>C(p.I382T)、c.739G>A(p.G247S)、c.1481C>T(p.T494M)、c.880G>C(p.G294R);我们的研究发现新的错义突变c.1279A>G(p.Y427H)及移码突变c.1128_1138del(p.Q377GfsTer12)。在本研究中,最常见的点突变为c.1145T>C(p.I382T),6 例患儿中3 例(50%)检出该位点突变(表2)。TH 蛋白由4个亚基组成,每个亚基含3个结构域:1.调控结构域,2.催化区域3.四聚体化结构域。本研究6例患儿存在的7 个突变位点均位于催化域(图1)。
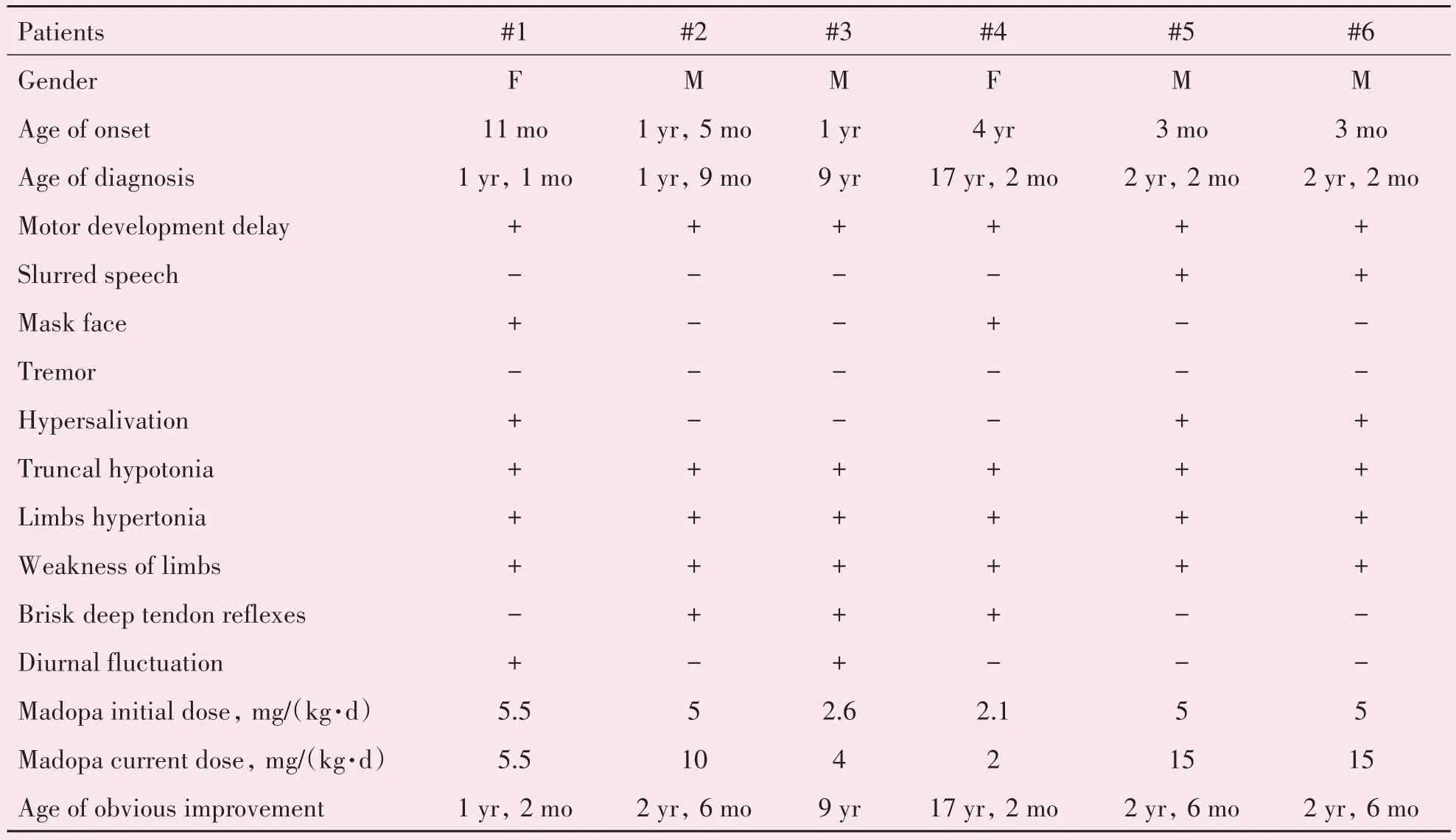
表1 6例THD患儿的临床表现、治疗和预后Table 1 Clinical manifestations,treatment and prognosis of 6 children with THD
2.4 治疗与随访
本研究的所有患儿均服用美多芭(左旋多巴/盐酸苄丝肼50 mg/200 mg 分散片,Madopa)治疗。根据患儿的临床表现和对副作用的耐受性进行调整。6例患儿开始美多芭治疗的中位数年龄为2岁2 个月(范围从13 个月到17 岁2 个月)。中位数治疗时间为1 年3 个月(范围从3 个月到3 年5 个月)。美多芭的初始剂量为2.1~5.5 mg/(kg·d)。目前美多芭的维持剂量为2~15 mg/(kg·d),分2~3 次服用。6 例患儿运动功能均显著改善。中位数随访时间为1 年3 个月(范围从3 个月到3 年5 个月)。5例患儿(#1,#2,#4,#5,#6)美多芭治疗后,能够独立行走的中位数时间为4 个月(范围从2 周到13 个月),#3 患儿(治疗前可独走,但姿势异常)在治疗开始后1 周,行走姿势恢复正常,随访中症状持续改善。6 例患儿韦氏智力测试结果均在正常范围,但患儿#2,#5,#6 韦氏智力测试中,语言理解能力分值低于平均水平(表1)。
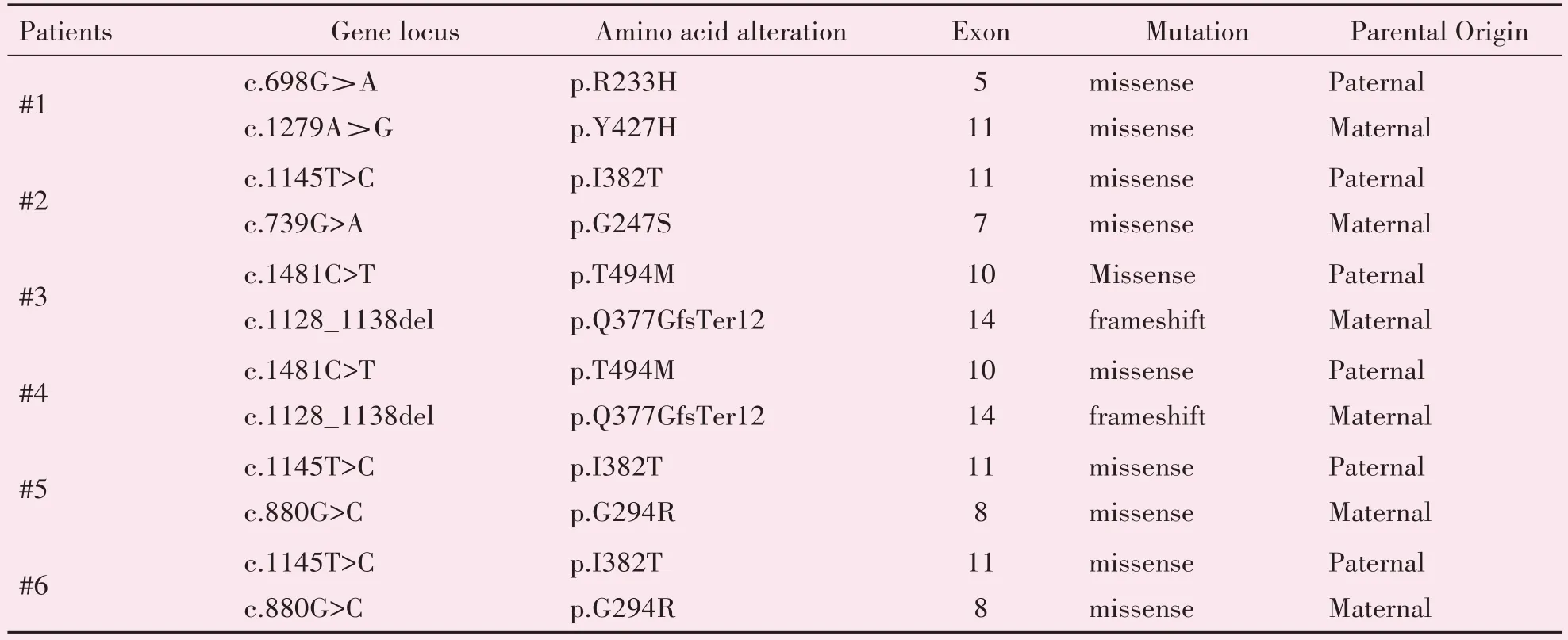
表2 6例酪氨酸羟化酶缺乏患儿的TH基因检测结果Table 2 TH gene test results of 6 children with tyrosine hydroxylase deficiency

图1 人类TH和本研究6例患儿突变位点的序列比对Fig.1 Sequence alignment of mutant sites between human TH and the 6 children in this study
3 讨论
DRD 是常染色体遗传运动障碍,是一种严重但可治疗的神经代谢性疾病,发生在儿童和青少年。其特征是进行性肌张力障碍,对低剂量左旋多巴治疗可有显著反应。DRD 的遗传模式有常染色体显性遗传、常染色体隐性遗传、散发,其中由GCH1基因突变引起的常染色体显性遗传的DRD最为常见。相比之下,TH基因突变引起THD 为常染色体隐性遗传,是DRD 罕见的发病机制,相较于GCH1基因突变引起的DRD,其症状更重,发病更早。
DRD 患儿女性居多,女性是男性患儿的2.5~4倍。GCH1基因突变引起的DRD 在女性患儿中的外显率是男性的2.3 倍[5],而Willemsen 等[6]对36 例THD 患儿的研究报道显示THD 患儿无明显性别差异。结合本研究的6 例THD 男女比例为4:2,提示THD并未呈现绝大多数DRD的女性优势比。
THD 多在生后数周到5 岁起病,常为下肢起病的部分性或全身性肌张力不全,最常见的症状是运动减少,躯干肌张力降低和肢体肌张力增高[7]。与既往文献报道的38.7%患儿有晨轻暮重表现相仿[8],本研究33.3%(2/6)的患儿有晨轻暮重表现。70%患儿在15 岁前即可出现严重症状[6]。根据症状的严重程度和对左旋多巴的反应性,THD 有3 种类型[4],分别为1 型:左旋多巴反应性肌张力不全(经典的DRD),2 型:婴儿帕金森症伴运动延迟,3型:进行性婴儿脑病。与1型THD患儿相比,2型及3型的运动及认知功能预后较差。本研究的6例患儿均属于1 型THD。THD 具有广泛的临床异质性,常被误诊为脑瘫或遗传性痉挛性截瘫,体格检查均可存在下肢腱反射活跃、肌张力增高等。THD也可能因为出现发作性肌张力障碍和异常行为而被误诊为癫痫。在本研究中,2 例患儿(#5,#6)既往在被误诊为脑瘫,2 例患儿(#3,#4)被误诊为遗传性痉挛性截瘫。与THD 相比,脑瘫是非进展性疾病,左旋多巴治疗无效,神经影像学可异常。而脑电图即可帮助鉴别癫痫与非癫痫性发作。此外,THD的晨轻暮重现象应与重症肌无力相鉴别,但前者新斯的明试验阴性,左旋多巴治疗有效,可予鉴别。总的来说,由于小剂量的左旋多巴可以使绝大多数THD 患儿明显改善,因此应考虑对肌张力障碍患儿进行左旋多巴试验,以免误诊。
酪氨酸羟化酶(tyrosine hydroxylase,TH)是儿茶酚胺(即多巴胺、去甲肾上腺素和肾上腺素)生物合成中的限速酶。TH 主要在脑和肾上腺表达。因此,通过获取组织样本进行酶学测定是不太可行的。THD 患儿脑脊液多巴胺代谢产物高香草酸和高香草酸与5-羟基吲哚乙酸比值降低,去甲肾上腺素代谢产物3-甲氧基-4-羟基苯基乙二醇降低,生物蝶呤和新蝶呤浓度正常[6]。然而,并不是所有的患儿都愿意接受侵入性检测,这突显了DRD 相关基因遗传分析的优势。TH基因位于染色体11p15.5,其包含14 个外显子,开放阅读框为1491bp,编码497 氨基酸。TH基因的纯合突变或复合杂合突变可引起THD。TH基因的错义突变是THD 的主要原因。截止至2020 年10 月,HGMD 数据库已经报道了88 个致病TH基因突变。其中c.698G>A(p.R233H)为热点突变[9]。Van den Heuvel等[10]报道了3例纯合突变p.R233H 患儿,症状符合2 型THD。Zhang[11]等亦报道了TH基因复合杂合突变的1例患儿,其存在1个p.R233H 突变,患儿亦表现为2 型THD。因此p.R233H 突变被预测与2型THD 有关。而本研究患儿#1 存在TH基因p.R233H 突变,但其临床表现为1 型THD。因此本研究结果提示p.R233H 突变并不能作为临床表型预测的指标。目前THD 基因型与表型之间的相关性尚未建立,但随着TH蛋白功能的进一步研究,这将成为可能。本研究6 例患儿均为TH基因的复合杂合突变引起的THD。6 例患儿共有7 个TH基因突变,其中2个是既往未见报道的新突变。患儿#1存在TH基因c.1279A>G 突变,导致氨基酸改变p.Y427H(该基因编码的第427 位密码子由酪氨酸变为组氨酸),为错义突变,患儿#3 及#4 存在TH基因c.1128_1138del 突变,导致氨基酸p.Q377GfsTer12(此序列变化导致第377 号氨基酸由谷氨酰胺变成甘氨酸,且下游第12 号氨基酸变为终止密码),为移码突变,突变位点在多物种比对具有高度保守性,不属于多态性位点,通过SIFT、PolyPhen、MutationTaster等软件均预测其致病,国内外均未见相同突变报道。患儿#3 及#4 虽来自同一家庭,基因突变位点一致,但两者在性别、起病年龄、临床表现、严重程度及疾病进程方面均存在差异,这也体现了该病的遗传异质性。另外有3例(50%)患儿存在相同TH基因突变位点,c.1145T>C(p.I382T)。该位点首次由台湾学者于2012 年报道[9]。本研究3 例患儿为继台湾学者报道后的再次报道。该位点是否呈现在中国人群高发趋势,有待更大样本的研究。
治疗方面,左旋多巴是治疗THD 的神奇药物。左旋多巴用量采用逐步滴定法。起始剂量通常为0.5~1 mg/(kg·d),每月增加0.1~0.5 mg/(kg·d),根据疗效及患儿对药物的耐受情况,维持剂量为3~10 mg/(kg·d),分1~3 次口服[12]。左旋多巴安全、有效,在剂量增加过快或过大时,可引起左旋多巴诱导的运动障碍。Couto 等[13]报道1 例患儿从起病到使用左旋多巴治疗相差19 年。患儿无法执行任何运动任务,借助轮椅15 年。左旋多巴治疗后,患儿有异常缓慢但持续的改善过程,笔者分析左旋多巴起效缓慢可能与该患儿长期的运动功能丧失相关。国内段丽芬等[14]报道1 例11 岁大患者,卧床11 年,经左旋多巴治疗取得扭转性效果,现已能基本正常生活,且副作用小。本研究患儿#4 虽然从起病到诊断明确,采用左旋多巴治疗,相差13年,其中完全卧床,生活不能自理长达4 年,但依然显示出对左旋多巴快速而显著的反应,而患儿#2规则服用美多芭治疗前,病程仅4 个月,但从服药到病情明显改善则经历近1 年时间,提示对药物反应的快慢,不完全由患儿病程决定,还可能与患儿的基因型,药物吸收情况等因素相关,这有待进一步探讨。尽管大多数患儿在左旋多巴启用后的2周即可表现疗效[6],但仍有少数患儿在数月内才有治疗反应(本研究患儿#2),因此,应耐心观察足够长的时间,以客观判断药物疗效,不可操之过急。
本研究存在一定局限性。首先,这是一项单中心、回顾性研究,可能造成选择偏倚。其次,研究纳入样本量较少,这与THD 为罕见病不无关系。今后我们计划开展多中心、大样本、前瞻性的研究,进一步总结THD患儿的临床和遗传特征。
综上所述,THD是一种罕见且严重的神经代谢疾病,其临床表现复杂多样,尽早识别和左旋多巴治疗,可明显改善预后。THD的诊断依赖于临床症状,左旋多巴诊断性治疗和TH基因的检测。本研究检测到TH基因2 个新的突变,错义突变c.1279A>G 及移码突变c.1128_1138del。本研究拓宽了THD 的基因型谱,为了解THD 的分子机制提供了新的思路。基因检测是THD 最重要的诊断方法,对后续治疗及遗传咨询有指导意义。
