18例经基因确诊的DYT1型肌张力障碍临床特点分析
2014-05-08王琳马凌燕杨英麦万新华
王琳 马凌燕 杨英麦 万新华
18例经基因确诊的DYT1型肌张力障碍临床特点分析
王琳 马凌燕 杨英麦 万新华
目的 分析DYT1型肌张力障碍患者的临床特点。方法 回顾性分析作者医院2009-06-2012-12收治的经基因检查证实的18例DYT1型肌张力障碍的临床资料,并与10例DYT6型肌张力障碍的临床特点进行比较。结果 18例DYT1型肌张力障碍患者中,男11例,女7例,起病年龄5~40岁,平均(15.6±9.0)岁,有8例患者有家族史。肢体(61.1%)是最常见的起病部位,上肢较下肢多见,其次是颈部(33.3%)。最常受累部位为上肢(36.8%)、颈部(24.6%)和下肢(22.8%),头面部几乎无影响,表现为节段型(50.0%)或全身型(38.9%)肌张力障碍。以肢体起病的患者平均起病年龄(11.5±5.1)岁,低于以颈部起病患者的(25.0±8.5)岁(< 0.01)。肢体起病的患者进展成为全身型肌张力障碍的比例(46.4%)高于以颈部起病的患者(16.7%)。与DYT6型肌张力障碍相比,DYT1型的起病年龄小于DYT6型(20.8岁)﹝(15.6±9.0)岁比(20.8±7.7)岁﹞,二者均以早发型肌张力障碍为主,颈部和上肢都是最初起病和最常受累的部位,但在起病部位上,DYT6型的患者以颈部起病更为多见(70.0%),主要影响颈部和上肢,躯干和下肢不受累。同时,颅段的症状如眼睑痉挛和构音不清发生率高,病变分布为节段型(60.0%)和局灶型(40.0%),没有全身型肌张力障碍。DYT1型和DYT6型肌张力障碍在病变累及部位和分布类型上的差异具有统计学意义(<0.01,=0.039)。结论 DYT1型肌张力障碍以早发节段型或全身型肌张力障碍为主要表现,肢体是最常见的起病部位,上肢较下肢多见,其次是颈部。上肢、颈部和下肢是最常受累的部位。DYT1型和DYT6型肌张力障碍在病变累及部位和分布类型上存在不同。
DYT1;DYT6;张力障碍;回顾性研究
肌张力障碍是一种不自主、持续性肌肉收缩引起的扭曲、重复运动或姿势异常的综合征。根据肌张力障碍的起病原因,可以分为原发性肌张力障碍、肌张力障碍叠加综合征、遗传变性病肌张力障碍、发作性肌张力障碍和继发性肌张力障碍[1]。其中原发性肌张力障碍没有明确的获得性病因,也不属于遗传变性病,肌张力障碍是临床上惟一的异常表现。分子生物学研究发现多数原发性肌张力障碍存在遗传学基础,以DYT1型肌张力障碍最为常见,绝大多数与编码扭转蛋白A(Torsin A)的TOR1A基因5号外显子的GAG序列缺失有关[2],其次是DYT6型肌张力障碍,与编码死亡相关蛋白1(THAP1)的T HAP1基因突变相关[3]。该研究回顾分析作者医院收治的经基因确诊的18例DYT1型肌张力障碍的临床特点,并与10例DYT6型肌张力障碍进行比较。
1 对象和方法
1.1 临床资料 所有病例均来自2009-06-2012-12中国医学科学院北京协和医院神经内科运动障碍病门诊,经基因筛查共发现18例DYT1型肌张力障碍和10例DYT6型肌张力障碍患者。行基因检查前均由患者本人或监护人签署书面知情同意书。
1.2 方法
1.2.1 临床资料收集:回顾性分析患者的性别构成、起病年龄、起病部位、病变分布、家族史等临床特点,并将DYT1型和DYT6型肌张力障碍患者的临床资料进行比较。依据中华医学会肌张力障碍诊断与治疗指南[1],按症状分布,将肌张力障碍分为局灶型、节段型、多灶型、全身型和偏身型。按发病年龄,将肌张力障碍分为早发型≤26岁和晚发型>26岁。起病部位和病变分布依照统一肌张力障碍评分量表(UDRS)中的病变部位进行判断,包括眼和上面部、下面部、下颌和舌、喉、颈部、肩部和近端上肢(左右)、远端上肢和手包括肘(左右)、骨盆和近端下肢(左右)、远端下肢和足包括膝(左右)、躯干共14个部位。
所有病例常规MRI检查正常,血清铜蓝蛋白水平正常。通过病史、查体和实验室检查确认所有病例为原发性肌张力障碍。
1.2.2 基因检查:所有病例进行TOR1A基因5号外显子GAG缺失突变筛查和THAP1基因的3个外显子及外显子内含子交界区测序。18例患者具有TORA1基因的3个碱基对缺失,确认为DYT1型肌张力障碍,均为c.904_906/907_ 909delGAG。有10例患者具有THAP1基因突变,诊为DYT6型肌张力障碍,其中2例突变位于1号外显子,6例位于2号外显子,2例位于3号外显子。
1.3 统计学处理 数据分析应用SPSS V20.0软件。计量资料经Kolmogorov-Smirnov检验均符合正态分布,以均值±标准差表示,采用 检验;计数资料用例表示,采用χ2检验。以<0.05为差异有统计学意义。
2 结果
DYT1型肌张力障碍患者中男11例,女7例,起病年龄5~40岁,平均(15.6±9.0)岁,其中早发型16例,晚发型2例,有8例患者有家族史。DYT6型肌张力障碍患者中男6例,女4例,起病年龄7~36岁,平均(20.8±7.7)岁,其中早发型9例,晚发型1例,有8例患者有家族史。DYT1型和DYT6型在起病年龄(=0.136)、性别构成(=0.954)和家族史(=0.069)间比较差异无统计学意义。两者在起病部位、受累部位和病变分布上的比较结果见表1~3。其中DYT1型肌张力障碍患者平均受累部位数为(3.2±1.6)个,DYT6型患者平均受累部位数为(2.2±1.1)个,两型间比较差异无统计学意义(=-1.669,=0.107)。
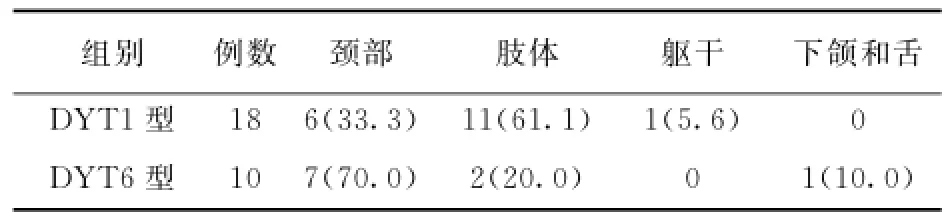
表1 DYT1型和DYT6型肌张力障碍起病部位比较 ﹝例(%)﹞

表2 DYT1型和DYT6型肌张力障碍受累部位比较 ﹝个(%)﹞

表3 DYT1型和DYT6型肌张力障碍患者分型分布 ﹝例(%)﹞
对DYT1型肌张力障碍的患者进行进一步分析,以肢体起病的患者平均起病年龄(11.5±5.1岁),低于以颈部肌张力障碍起病的患者﹝(25.0± 8.5)岁;=4.142,<0.01﹞。其中7例以上肢起病的患者起病年龄(10.1±3.5)岁,4例以下肢起病的患者发病年龄(13.8±7.1)岁,二者间比较无统计学差异(=-1.155,=0.278)。肢体起病的患者约半数(46.4%)进展成为全身型肌张力障碍,高于以颈部起病的患者(16.7%;χ2=1.409,=0.235)。≤10岁起病的患者中全身型肌张力障碍的比例为60.0%,10~20岁起病的患者为42.9%,>20岁起病的患者只有16.7%进展为全身型肌张力障碍。
3 讨论
1989年Ozelius等首次在一个非德系犹太家系中发现原发性肌张力障碍的致病基因位于9号染色体长臂[4],1997年将其最终定位于9q34,命名为TOR1A基因,编码扭转蛋白A,与TOR1 A基因异常相关的原发性肌张力障碍命名为DYT1型肌张力障碍[1]。TOR1A基因5号外显子3个碱基对缺失(c.904_906/907_909delGAG)[5]是最常见的突变形式,约占99%以上,导致扭转蛋白A的羧基末端302/303位点谷氨酸缺失。其他类型的突变也有报道,包括c.863G>A[6]、c.613T>A[7]、c.934_937del AGAG[8]、c.966_983del[9]等,所占比例不到1%。
DYT1型肌张力障碍以犹太人种发病率最高,为1/1.6万~1/2万,非犹太人种发病率约为1/ 20万[10]。在德系犹太人种患者中,典型的表现是青少年期以单个肢体起病,下肢较上肢更为常见且起病年龄更早,病情逐渐进展,约65%的患者经5~10年发展为全身型肌张力障碍[11]。但随着临床病例的积累,很多学者发现不同种族之间, DYT1型肌张力障碍的表型存在差异[12]。
该组DYT1型肌张力障碍患者以肢体(以上肢为主)为最常见起病部位,其次为颈部;在病变分布上,节段性肌张力障碍是最常见的类型(50%),另有1/3的患者为全身型,少数患者是局灶型;上肢是最常受累的部位,其次颈部、下肢和躯干;与颈部起病的患者相比,肢体起病的患者起病年龄小,进展为全身型肌张力障碍的可能性大;随着起病年龄的增高,全身型肌张力障碍的比例降低。
结合国内2篇文献报道的经基因检查确诊的8例患者[13-14],可总结出我国DYT1型肌张力障碍患者的临床特点:平均起病年龄14.5岁,肢体起病最多见,但颈部起病也常见,无颅段起病;全身型和节段型分布的比例相当,局灶型少见,无多灶型分布;上肢是最常见受累部位,其次是颈部和下肢。与国外文献进行比较发现,起病年龄大于西方国家(徳系犹太人种10.8岁,非徳系犹太人种13.0岁)[12],颈段起病和受累更为突出,上肢起病较下肢常见,节段型肌张力障碍的比例也明显高于西方国家[15-17]。
DYT1和DYT6型肌张力障碍具有很多共同点:二者同为早发型肌张力障碍;男性患者稍多于女性;颈部和上肢都是最初起病和最易受累的部位。目前研究发现,基因突变可以破坏TOR1 A基因启动子与THAP基因的相互作用,提示不同亚型的原发性肌张力障碍可能存在共同致病通路[18]。但结合分析该组患者临床资料发现两者也存在很多不同:(1)DYT1和DYT6型肌张力障碍的平均起病年龄分别为(15.6±9.0)岁和(20.8± 7.7)岁,前者小于后者;(2)在DYT1型患者中具有家族史的比例(44.4%)低于DYT6型(80.0%),可能与DYT1型的外显率(30%)[19]低于DYT6型(57%)[20]有关;(3)在起病部位上, DYT1型以肢体起病为主,也有部分患者是颈部起病,而DYT6型的患者以颈部起病为主,但二者间比较差异无统计学意义;(4)在病变累及部位间比较差异具有统计学意义,DYT1型累及颈部、肢体和躯干,头面部几乎无影响,而DYT6型主要影响颈部和上肢,躯干和下肢不受累,颅段的症状如眼睑痉挛和构音不清的高发生率很值得关注。此外DYT1型患者的病变受累部位多于DYT6型﹝(3.2±1.6)个vs.(2.2±1.1)个﹞;(5)在病变分布类型上二者也具有统计学差异,DYT1型患者表现为全身型或节段型较多,DYT6型则以节段型和局灶型为主,没有全身型。
DYT1型和DYT6型肌张力障碍致病基因的发现,提高了人们对原发性肌张力障碍的认识。尽管二者在起病年龄、起病部位和疾病分布上存在差异,但临床表现有很多重叠之处,因此仅仅从临床表现上难以将二者准确地区分,基因检查对于明确诊断很有帮助。对于早发型肌张力障碍的患者,特别是节段型或全身型分布,或是有家族史的患者,不仅要进行DYT1型TOR1A的基因检查,也应该进行DYT6型THAP1的基因检查,有助于疾病的早期诊断。
[1]中华医学会神经病学分会帕金森病及运动障碍学组.肌张力障碍诊断与治疗指南[J].中华神经科杂志,2008,8:570-573.
[2]Ozelius LJ,Hewett JW,Page CE,et al.The early-onset torsion dystonia gene(DYT1)encodes an ATP-binding protein[J].Nat Genet,1997,17:40-48.
[3]Fuchs T,Gavarini S,Saunders-Pullman R,et al.Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia[J].Nat Genet,2009,41:286-288.
[4]Ozelius LJ,Kramer PL,Moskowitz CB,et a1.Human gene for torsion dystonia located on chromosome 9q32-34[J]. Neuron,1989,2:1427-1434.
[5]Warner TT,Jarman P.The molecular genetics of the dystonias[J].J Neurol Neurosurg Psychiatry,1998:64:427-429.
[6]Zirn B,Grundmann K,Huppke P,et al.Novel TOR1A mutation p.Arg288Gln in early onset dystonia(DYT1)[J].J Neurol Neurosurg Psychiatry,2008,79:1327-1330.
[7]Calakos N,Patel VD,Gottron M,et al.Functional evidence implicating a novel TOR1A mutation in idiopathic,late onset focal dystonia[J].J Med Genet,2010,47:646-650.
[8]Kabakci K,Hedrich K,Leung JC,et al.Mutations in DYT1:extension of the phenotypic and mutational spectrum[J]. Neurology,2004,62:395-400.
[9]Leung JC,Klein C,Friedman J,et al.Novel mutation in the TOR1A(DYT1)gene in atypical early onset dystonia and polymorphisms in dystonia and early onset parkinsonism[J]. Neurogenetics,2001,3:133-143.
[10]Muüller U.Non-manifesting carriers of the DYT1 dystonia mutation[J].Neurology,2005,64:347-349.
[11]Risch N,de Leon D,Ozelius L,et al.Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population[J].Nat Genet, 1995,9:152-159.
[12]Lee WW,Ahn TB,Chung SJ,et al.Phenotypic differences in Dyt1 between ethnic groups[J].Curr Neurol Neurosci Rep,2012,12:341-347.
[13]Zhang SS,Fang DF,Hu XH,et al.Clinical feature and DYT1 mutation screening in primary dystonia patients from South-West China[J].Eur J Neurol,2010,17:846-851.
[14]Yang JF,Wu T,Li JY,et al.DYT1 mutations in early onset primary torsion dystonia and Parkinson disease patients in Chinese populations[J].Neurosci Lett,2009,450:117-121.
[15]Bressman SB,Sabatti C,Raymond D,et al.The DYT1 phenotype and guidelines for diagnostic testing[J].Neurology, 2000,54:1746-1752.
[16]Anca MH,Zaccai TF,Badarna S,et al.Natural history of Oppenheim’s dystonia(DYT1)in Israel[J].J Child Neurol, 2003,18:325-330.
[17]Kramer PL,Heiman GA,Gasser T,et al.The DYT1 gene on 9q34 is responsible for most cases of early limb-onset idiopathic torsion dystonia in non-Jews[J].Am J Hum Genet, 1994,55:468-475.
[18]Gavarini S,Cayrol C,Fuchs T,et al.Direct interaction between causative genes of DYT1 and DYT6 primary dystonia[J].Ann Neurol,2010,68:549-553.
[19]Bressman SB,de Leon D,Brin MF,et al.Idiopathic dystonia among Ashkenazi Jews:evidence for autosomal dominant inheritance[J].Ann Neurol,1989,26:612-620.
[20]Saunders-Pullman R,Raymond D,Senthil G,et al.Narrowing the DYT6 dystonia region and evidence for locus heterogeneity in the Amish-Mennonites[J].Am J Med Genet A, 2007,143A:2098-2105.
WAN Xin-hua,Email:dr.wanxh@gmail.com
Objective To summarize the clinical features of DYT1 dystonia.Methods Clinical data of 18 patients with DYT1 dystonia treated in our hospital from June 2009 to December 2012 were retrospectively reviewed,and compared with the clinical data of 10 patients with DYT6 dystonia.Results Eighteen DYT6 dystonia patients were recruited,included 11 males and 7 females with onset age ranging 5-40 years old,the mean age of onset was(15.6±9.0)years old.Eight patients had a positive family history.The most common site of onset was limb(61.1%),upper extremity was predominant,next was neck(33.3%).The frequently affected anatomical sites were arm(36.8%),neck(24.6%),and leg(22.8%),with face almostly spared.Most patients presented with segmental(50.0%)or general(38.9%)dystonia subtypes.The onset age of the patients with limb-onset dystonia was significantly younger than that of the patients with neck-onset dystonia[(11.5± 5.1)years.(25.0±8.5)years,<0.01].Limb-onset dystonia became generalized(46.4%)more frequently than neck-onset dystonia(16.7%).The mean onset age of DYT6 was(20.8±7.7)years old,older than that of DYT1[(15.6±9.0)years old].Both of the two types of dystonia mainly presented as early-onset dystonia,neck,and upper extremity were the initial affected and common affected sites,but neck was the main site in DYT6,which was different from DYT1.In DYT 6,neck and upper extremity were mainly affected,trunk and lower extremity were not affected at all,and there were high incidence of dystonia in eye,jaw,and tongue.Segmental(60%)and focal(40%)subtypes were the main forms of dystonia,general dystonia was absent.The differences in site involved and clinical subtype between DYT1 and DYT6 were statistically significant(<0.01,=0.039).Conclusions Early-onset segmental or general dystonia is the main manifestation of DYT1 dystonia. The most common site of onset is limb,upper extremity was predominant,followed by neck.The most frequently affected site is upper extremity,next come neck and lower extremity.DYT1 and DYT6 have different sites involved and clinical subtypes.
DYT1;DYT6;dystonic disorders;retrospective study
R746.9
:A
:1006-2963(2014)05-0352-04

2014-01-26)
(本文编辑:时秋宽)
10.3969/j.issn.1006-2963.2014.05.012
100730中国医学科学院北京协和医学院北京协和医院神经内科(王琳、杨英麦、万新华);100050首都医科大学附属北京天坛医院神经内科(马凌燕)
万新华,Email:dr.wanxh@gmail.com
