复合型露酒在不同浸泡条件下主要活性成分及挥发性物质变化研究
2021-04-11朱定国程宏连
朱定国,王 广,程宏连,程 超
(1.来凤县中等职业技术学校,湖北来凤 445700;2.湖北民族大学生物科学与技术学院,湖北恩施 445000;3.重庆市酒类管理协会,重庆 404100)
中国除了茶饮之外,大概再没有一种液体能够像酒一样让人嗜之若命,也再没有一样食品能够像中国酒一样,与中华上下五千年文化一样源远流长。随着社会的不断前进、发展,中国酒也在不断地适应新的变化。从发酵酒到蒸馏酒再到露酒,从液态法发酵到固态法发酵,从高度化、烈性化、大众化到低度化、健康化、定制化等等,无不反映着中国酒的发展历程。在这一系列变化中尤以露酒最为明显,露酒作为中国酒里面重要的一种,有着举足轻重的地位。本文的复合型露酒主要以绵柔型小曲白酒为基酒[1],桂花、绞股蓝、藤茶为主要浸泡物,它不仅具备基酒所含有的酸、酯、醛、醇等多种物质[2-3],并且具有扩张血管、降低颅内压力、降低胆固醇、预防心血管疾病等药理功效[4-7],此外被浸泡物亦具备舒适的挥发油香气[8-10],富含黄酮、多酚、皂甙、多糖等[11-15],具有抗氧化[16-18],降血糖、血脂[19-21],延缓衰老[22-24],抗菌、消炎等诸多功效[25-28],以及其他营养功能。
1 材料与方法
1.1 材料、试剂及仪器
样品:绵柔型小曲白酒,恩施巴王酒业有限公司;藤茶(优级),湖北仙芝堂生物科技有限公司;绞股蓝(优级),湖北仙芝堂生物科技有限公司;桂花(优级),湖北仙芝堂生物科技有限公司。
试剂及耗材:无水乙醇(AR 级),国药集团化学试剂有限公司;正丁醇(AR 级),国药集团化学试剂有限公司;高氯酸(AR级),国药集团化学试剂有限公司;盐酸(AR 级),国药集团化学试剂有限公司;氢氧化钠(AR级),国药集团化学试剂有限公司;乙酸(AR 级),国药集团化学试剂有限公司;香草醛(AR 级),国药集团化学试剂有限公司;福林酚试剂标准品(色谱级),国药集团化学试剂有限公司;(一水)没食子酸标准品(色谱级),国药集团化学试剂有限公司;齐墩果酸标准品(色谱级),国药集团化学试剂有限公司。
仪器设备:台式分光测色仪(CS-820N),杭州彩谱科技有限公司;紫外可见分光光度计(UV-9000S),上海元析仪器有限公司;杯式超声波细胞破碎机,宁波新芝生物科技股份有限公司;Flavour-Spec®风味分析仪,山东海能科学仪器有限公司;旋转蒸发仪(BC—R206),上海贝凯生物化工设备有限公司;恒温水浴锅(SG),上海苏豪智能系统有限公司。
1.2 试验方法
1.2.1 浸泡时间对复合露酒活性成分及色度的影响
浸泡时间分别为1 d、5 d、10 d、20 d、30 d、40 d、50 d、60 d、120 d,酒精度45.00 %vol,料液比3.00 %,测定活性成分(总糖、多酚、黄酮、皂甙)及色度。
1.2.2 酒精度对复合露酒活性成分及色度的影响
酒精度分别为15.00 %vol、25.00 %vol、35.00 %vol、45.00 %vol、55.00 %vol,料液比为3.00%,浸泡时间30 d,测定活性成分(总糖、多酚、黄酮、皂甙)及色度。
1.2.3 料液比对复合露酒活性成分及色度的影响
料液比分别为2.00%、3.00%、4.00%、5.00%、6.00 %,酒精度45.00 %vol,浸泡时间30 d,测定活性成分(总糖、多酚、黄酮、皂甙)及色度。
1.2.4 超声波处理对复合露酒活性成分及色度的影响
测定超声波前处理影响时采用酒精度为45.00 %vol,料液比为3.00 %(m/v),处理功率为400 W,单次处理时间50 s,间隔时间60 s,处理次数分别为1 次、10 次、20 次、30 次、40 次、50 次,测定活性成分(总糖、多酚、黄酮、皂甙)及色度,超声处理在水浴中进行。
1.2.5 活性成分测定
1.2.5.1 黄酮的测定方法:Al(NO3)3-NaNO2-NaOH法[29]
芦丁标准曲线绘制:精确称取0.02 g 芦丁于洁净烧杯中,用70.00 mL 的30%乙醇分两次溶解,转移于100 mL 容量瓶中,同时用30 %乙醇定容,备用。取5.00 %的亚硝酸钠0.30 mL 于7 支带塞试管,并分别依次加入配制好的芦丁溶液0.00 mL、0.50 mL、1.50 mL、2.00 mL、2.50 mL、3.00 mL,摇匀、静置6 min后,依次加入0.30 mL浓度为10%的硝酸铝溶液,摇匀静置6 min,然后依次加入4.00 mL 浓度4%的氢氧化钠,并用30%乙醇溶液定容10 mL,摇匀静置10 min 后,于510 nm 处测定吸光度,并以芦丁浓度(mg/mL)为横坐标,以A510为纵坐标绘制标准曲线,拟合回归方程为y=10.286x+0.0024,R2=0.9997。
样品中黄酮含量测定:取露酒上清液10.00 mL于洁净烧杯中,加入450.00 mL 30 %乙醇,搅拌均匀,并于500 mL容量瓶中用30%乙醇溶液定容(即稀释50 倍),取1 mL 样品稀释液,并按照标准曲线测定方法操作,按照公式(1)计算样品中黄酮含量。

1.2.5.2 多酚含量测定:Folin-Ciocalteu法[30]
没食子酸标准曲线绘制:取7支洁净带塞的玻璃试管,分别加入5.00 mL超纯水、2.50 mL福林-酚试剂,然后依次加入0.00、0.25 mL、0.50 mL、0.75 mL、1.00 mL、1.25 mL、1.50 mL 的没食子酸,摇匀静置30 s 后加入12 %浓度的碳酸钠4.00 mL,最后用超纯水定容到25 mL,于水浴锅中45 ℃保温90 min,之后在760 nm 处测定其吸光度,并以没食子酸浓度(mg/mL)为横坐标,以A760为纵坐标绘制标准曲线,拟合回归方程为y=13.821x+0.0002,R2=0.9992。
样品中总酚的测定:取露酒上清液1.00 mL 于50 mL 容量瓶中,用超纯水定容(即样品稀释50倍),并按照标准曲线测定方法操作,按照公式(2)计算样品中总酚含量。

1.2.5.3 皂甙的测定:齐墩果酸法[31]
齐墩果酸标准曲线绘制:精确称取0.02 g 的齐墩果酸,用甲醇溶液溶解并定容到50 mL(即齐墩果酸的浓度为0.40 mg/mL),取7 支带塞的洁净试管,依次加入0.00、0.05 mL、0.10 mL、0.15 mL、0.20 mL、0.25 mL、0.30 mL 的0.40 mg/mL 齐墩果酸溶液,然后在70 ℃温度下水浴蒸干,然后依次加入新配制的5 %香草醛-冰醋酸溶液0.20 mL,高氯酸0.80 mL,于60 ℃下水浴15 min,再加入冰醋酸5.00 mL,摇匀后在550 nm 处测定其吸光度,以齐墩果酸浓度(mg/mL)为横坐标,以A550为纵坐标绘制标准曲线,拟合回归方程为y=7.8946x-0.0273,R2=0.9998。
样品皂甙测定:取露酒上清液10.00 mL并加入无水乙醇40.00 mL,在旋转蒸发仪上进行水浴加热至蒸干,然后加入超纯水10.00 mL、30.00 mL 正丁醇,萃取4 次,将萃取液合并减压蒸干,用甲醇定容到10 mL,取50 μL(稀释200 倍)按照标准曲线测定步骤操作,按照公式(3)计算样品中皂甙含量。

1.2.5.4 总糖的测定:3,5-二硝基水杨酸法[32]
标准曲线的绘制:准确称量1.00 g 的葡萄糖,用超纯水进行溶解,并于1000 mL 容量瓶中定容,即浓度为1.00 g/L。取7 支洁净带塞的试管,依次加入配制好的葡萄糖溶液0.00、0.20 mL、0.40 mL、0.60 mL、0.80 mL、1.00 mL、1.20 mL,并补加超纯水到总量为2.00 mL,每支试管加入4.00 mL 的3,5-二硝基水杨酸溶液,摇匀,沸水浴5 min,取出冷却,在540 nm 处测定其吸光值并绘制标准曲线,并以葡萄糖浓度(mg/mL)为横坐标,以A540为纵坐标绘制标准曲线,拟合回归方程为y=1.2621x-0.0089,R2=0.9995。
样品测定:取10.00 mL复合露酒上清液于100 mL容量瓶中(通过预实验控制总糖在0.3 g 左右),加入浓度为6 mol/L 的盐酸溶液5.00 mL,超纯水20.00 mL,摇匀。然后在68 ℃的水浴锅中进行水解15 min,取出冷却,用浓度为6 mol/L 的氢氧化钠调至中性,并定容到100 mL 备用。取2.00 mL 备用液按照标准曲线测定操作(稀释50 倍),按照公式(4)计算样品中总糖含量。

1.2.5.5 色度测定
先取10.00 mL 无水乙醇于台式分光测色仪(CS-820N)的比色皿中,并用专用的擦拭纸将透光面擦拭干净,放置于已经预热的仪器中进行调零操作,然后取10.00 mL的复合露酒上清液按照同样的步骤测定样品的色度值(包含L 值、a 值、b 值)并绘制曲线图。
1.2.6 复合露酒的风味测定方法
将不同浸泡时间组的露酒取样10 mL 冷藏备用,待整体露酒原液酒样取齐后进行挥发性风味测定(即分别浸泡0、5 d、10 d、20 d、30 d、40 d、50 d、60 d),每组3个平行。
测定方法及实验条件为:将露酒原液样品稀释10 倍后取1 mL 置于20 mL 顶空瓶中,60℃孵育15 min 后进样200 μL。其中气相-离子迁移谱单元条件为:选择WAX 30 m ID:0.53 mm 的色谱柱,以N2为载气,IMS 温度45 ℃,柱温60 ℃,分析时间40 min;自动顶空进样单元条件为:进样体积为200 μL,进样针温度85 ℃,孵育时间为15 min,孵育温度为60 ℃,孵化转速为500 r/min。
2 结果与分析
2.1 浸泡时间对复合露酒活性成分及色度的影响
复合露酒主要活性成分黄酮、总酚、总糖、皂甙的含量及色度变化趋势如图1、图2、图3所示。

图1 浸泡时间对复合露酒黄酮与总酚的影响

图2 浸泡时间对复合露酒总糖与皂甙的影响

图3 浸泡时间对复合露酒色度的影响
由图1 可以看出,黄酮和多酚的浸出量在30 d时达到最大值,之后开始逐步下降,可能是由于黄酮为醇溶性物质,前期由于基酒中黄酮含量少,所以其溶出速率快,后期由于浸泡时间的延长,其溶出速率逐渐降低,此外在后期浸泡过程中,已经浸出的黄酮和多酚可能会被空气氧化导致其含量有所下降。从图2 可看出,总糖和皂甙含量变化趋势相似,在20 d 时达到最大值,之后出现交替下降和上升,但总体趋势为在浸泡20 d 后复合露酒浸泡液中总糖和皂甙含量下降。
从图3 和表1 可以看出,随着浸泡时间的延长,复合露酒的L 值显著下降,a 和b 值显著上升。露酒在浸泡过程中其色度L 值不断降低,即露酒的颜色变得越来越暗(深),这与浸泡过程中各种植物性的活性成分溶出有很大的关系。色度a*值表示的是红色,随着数值的不断增加红色加深,b*值表示的是黄色,随着数值增加黄色加深,而复合露酒色度值a*和b*随浸泡时间延长逐渐增加,说明二者复合颜色即橙红色在不断加深。这可能是由于随着浸泡时间延长,黄酮、多酚、皂甙等物质溶出量增多,且部分被氧化从而使露酒颜色加深。

表1 不同浸泡时间复合露酒活性成分及色度差异显著性
2.2 料液比对复合露酒活性成分及色度的影响
不同总料液比的实验过程中,复合露酒的主要活性成分黄酮、总酚、总糖、皂甙的含量及色度变化趋势如图4、图5、图6所示。

图4 料液比对复合露酒黄酮及总酚的影响

图5 料液比对复合露酒总糖及皂甙的影响

图6 料液比对复合露酒色度的影响
实验结果显示,基酒为45.00 %vol,浸泡30 d,在桂花∶绞股蓝∶藤茶=3∶2∶1 比例不变情况下,随物料浓度的增加总黄酮、总酚、总糖、皂甙含量均显著上升,但当物料浓度大于6.00 %后,浸泡的酒体口感苦涩,难以下咽,故最大料液比只用到6.00%;在不同料液比情况下,该露酒色度值a*和色度值b*变化不大,而色度值L 在5.00 %(m/v)时候达到最大值。
2.3 酒精度对复合露酒活性成分及色度的影响
当物料浓度为3.00 %,浸泡时间30 d,复合露酒的生物活性成分、色度与基酒的酒精度关系如图7、图8、图9所示。

图7 酒精度对复合露酒黄酮及总酚的影响

图8 酒精度对复合露酒总糖及皂甙的影响
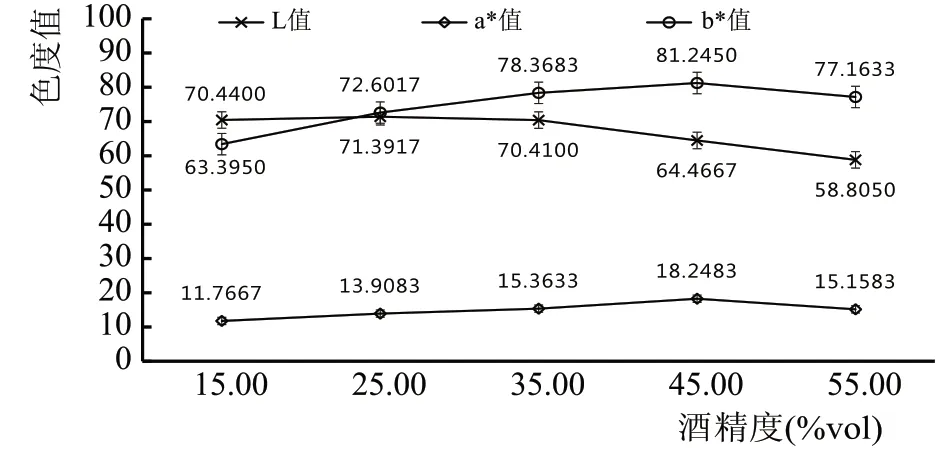
图9 酒精度对复合露酒色度的影响
从图7—图9 可看出,随基酒酒精度上升,黄酮、多酚、皂甙和多糖含量逐步增加,但多酚类物质在基酒酒精度超过45.00%vol 时,浸出量有轻微下降。主要由于黄酮、多酚、皂甙均为醇溶性,因此随基酒酒精含量上升,浸出量也随着上升,此外总糖含量上升可能是由于桂花、绞股蓝、藤茶等材料中含有一些小分子糖类物质,能够溶解于基酒中。而多酚类物质由于氧化程度较其他几种活性成分严重,故在酒精度超过45.00 %vol 后,总多酚含量是下降趋势。在整个浸泡过程中,露酒色度L 值随酒精度增加而减小(即变暗),a*和b*值随酒精度增加是先增高后降低,主要原因是由于酒精度增加使黄酮、多酚、总糖、皂甙等溶出量逐渐增加,到后期物料中活性成分溶出和被消耗(氧化等)达到近似平衡。
2.4 超声前处理对复合露酒活性成分及色度的影响
通过前期资料[33-35]查询采取固定超声处理功率、处理时间及两次处理之间的间隔时间(即超声功率为400 W,处理时间为50 s,处理间隔为60 s),采取不同超声次数处理,并取静置1 d 后的上清液测其主要活性成分及色度变化情况,结果见图10、图11、图12。

图10 超声次数对复合露酒黄酮及总酚的影响
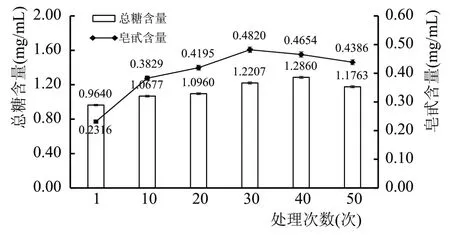
图11 超声次数对复合露酒总糖及皂甙的影响

图12 超声次数对复合露酒色度的影响
通过图10—图12 发现,随超声次数的不断增加,黄酮、多酚、皂甙、总糖的溶出量呈现先增加后减少的趋势,但各种活性成分达到最高值的超声次数不同,如黄酮类物质在超声处理20 次时,浸出量达到峰值,而皂甙和总酚均在超声次数为30 次时达到峰值,总糖在超声处理40 次时达到峰值。通过对比同等条件但未进行超声处理的数据,可以发现在超声处理次数为20~30 次的情况下,复合型露酒的4 种主要活性成分含量显著提高,尤其是对于总酚和皂甙含量的溶出效率,同样浸泡时间情况下,浸出率提高10~15 倍,这对于产业化生产能显著缩短露酒原酒制作(浸泡)周期,节约资金、提高资金周转率。可能是由于20~30 次超声处理能够有效破碎细胞,释放细胞内容物,进而达到增加露酒内4 种活性成分含量的作用,但是随超声次数增加,超声波处理对生物活性成分的分子结构造成破坏,使得测量结果偏低。
在不同超声次数的作用下,复合型露酒的色度变化趋势与活性成分的变化呈现很强的相关性,随着活性成分浸出量的逐渐增加,L 值逐渐下降,a*值和b*值逐渐上升,在超声次数为20~40 次时基本上达到峰值。相对于确定时间的浸泡实验(30 d,料液比为3.00%,酒精度为45.00%vol),露酒的颜色能够更快达到稳定,且整体颜色更偏深褐色一些,分析原因可能是随着超声处理次数的增加,引起浸泡原料的细胞破裂的同时造成局部温度过高,导致一部分大分子活性成分释放的同时也被加速氧化,从而显现出褐色。
2.5 不同浸泡时间复合露酒的挥发性物质分析
2.5.1 不同浸泡时间复合露酒挥发性物质
直接对比不同浸泡时间下露酒原液中的挥发性有机物差异,其3D图如图13所示(横坐标为不同浸泡时间下的样品,纵坐标为保留时间的峰强度)。

图13 不同浸泡时间复合露酒原液挥发性物质3D图
由图13 可发现,不同浸泡时间复合露酒原液,其挥发性成分形成的3D 图整体的相似性很高,但是在部分保留时间段上可以直观的看出不同浸泡时间条件下的露酒原液有细微区别,为了更好的分析对比,采取俯视图进行详细分析。
不同浸泡时间露酒原液挥发性物质的俯视图如图14 和图15 所示,其中纵坐标代表气相色谱的保留时间,横坐标代表离子迁移时间,整个图背景为蓝色,横坐标1.0 处红色竖线为RIP 峰(反应离子峰,经归一化处理),RIP 峰两侧的每一个点代表一种挥发性有机物。颜色代表物质的浓度,白色表示浓度较低,红色表示浓度较高,颜色越深表示浓度越大。

图14 不同浸泡条件下的露酒原液挥发性物质俯视图

图15 不同浸泡条件下的露酒原液挥发性物质俯视图(扣除0 d)
从图14—图15 可直观地看出,不同浸泡时间复合露酒的挥发性有机物差异较小。为了更加明显比较不同浸泡时间样品间的差异,采用差异对比模式,即选取其中一个浸泡时间下样品谱图作为参比,其他样品谱图扣减参比。如果二者挥发性有机物一致,则扣减后的背景为白色,而红色代表该物质的浓度高于参比,蓝色代表该物质的浓度低于参比(本次主要研究不同浸泡时间下的露酒原液挥发性物质差异性,故实验选择0 d 的测定组作为参比数据)。随浸泡时间延长,露酒中挥发性成分变化较大,且呈现一定的规律性,其中发现0、5 d、20 d的气相离子迁移谱图差异较小,相似度较高,而10 d、30 d、40 d 三者的相似性较高,组间差异较小,50 d、60 d 的组间差异也较小,另外浸泡时间越长,样品差异就越大。而对于所有样品而言,均是呈现随分离时间延长,样品挥发性物质越来越少的规律,即表明该露酒原液的主要挥发性物质为低沸点、易挥发的化合物,这一点与露酒的主要香味来源有关,即该露酒的香气是以桂花为主,辅以藤茶、绞股蓝的香味,在绵柔型白酒的携带作用下的一种复合型香味,而这一结论与前期有关研究也是一致的[36-38]。
2.5.2 不同浸泡时间复合露酒原液指纹图谱
如图16 和图17 所示,其中图中每3 行代表一个露酒样品中选取的全部信号峰(3 组平行试验),每一列代表同一挥发性有机物在不同浸泡时间下露酒样品中的信号峰。

图16 不同浸泡时间下露酒的挥发性有机物的Gallery Plot图(全部)

图17 露酒的挥发性有机物的Gallery Plot图(0 d、5 d、20 d、40 d和60 d)
从图16—图17 可发现,不同浸泡时间露酒的挥发性有机物指纹谱图,有如下规律:0、5 d、20 d、40 d、60 d相似,10 d、30 d、50 d较相似,造成此现象的原因极有可能是由于浸泡过程中露酒的各种生物活性成分变化(结合、分解等),造成露酒原液中成分周期性变化(这一点可以从0~60 d 浸泡下测定的总酚与总糖含量的周期性变化推测,尤其是0、5 d、20 d、40 d、60 d 的时候总糖含量均在拐点处),从而影响露酒整体的蒸气压所造成的,这一点在露酒原液的口感变化上得到证实且与果酒方面的研究吻合[39]。
从图16—图17 中还发现,A 区域表明露酒中部分挥发性有机物的含量随浸泡时间延长变化较小,主要有:乙醇、丁醇、丁酸乙酯、3-甲基丁醇和2-甲基-1-丙醇等;B 区域和E 区域表明露酒中部分挥发性有机物的含量随浸泡时间延长逐渐升高,主要有:己醇、2,6-二甲基吡嗪和乙酸乙酯等;C 区域表明0、5 d、20 d、40 d、60 d 露酒中特征挥发性有机物,其浓度高于10 d、30 d、50 d 露酒;D 区域表明10 d、30 d、50 d 露酒中特征挥发性有机物,其浓度高于0、5 d、20 d、40 d、60 d 露酒,即整体上可以看出每种样品的完整挥发物信息以及样品之间挥发性有机物的差异。
从图17 中可以发现以下规律:F 区域表明露酒中部分挥发性有机物的含量随浸泡时间延长逐渐降低,主要有:乙酸异戊酯和2-丁酮等;G区域表明露酒中部分挥发性有机物的含量随浸泡时间延长变化较小,主要有乙醇、丁酸乙酯、2-甲基-1-丙醇、3-甲基丁醇和丁醇等;H 区域表明0、5 d、20 d、40 d 和60 d 露酒中部分挥发性有机物的含量随浸泡时间延长逐渐升高,主要有:己醇、乙酸乙酯、己酸乙酯、2,6-二甲基吡嗪、戊酸乙酯、2-甲基丁酸乙酯和3-甲基丁酸乙酯等,同时60 d 露酒的风味物质的浓度最高;I 区域表明20 d 露酒的特征挥发性有机物,其浓度高于其他露酒。
2.5.3 不同浸泡时间下露酒原液的动态主成分分析聚类
如图18、图19 所示,其中样品相近则代表差异小,相隔远则代表组分差异明显。
通过图18、图19 发现,不同浸泡时间复合露酒其挥发性物质具有一定相似性,大致上可以分为3个特征组,其中0、5 d、20 d 浸泡组的距离较近,相似度高;40 d、60 d 浸泡组相似度高;30 d、50 d 浸泡组的相似度较高;同时0、5 d、20 d 浸泡组与40 d、60 d 浸泡组的相似度要高于与30 d、50 d 浸泡组的相似度。不同浸泡时间下的露酒原液特征成分分析结论是与Gallery Plot图结论一致的,另外特征成分分析图还有利于辅助后期成品的浸泡时间的优化。
2.5.4 不同浸泡时间复合露酒的气相离子迁移色谱

图18 不同浸泡时间复合露酒的聚类分析

图19 不同浸泡时间下露酒样品的聚类分析(0 d、5 d、20 d、40 d和60 d)
0、5 d、10 d、20 d、30 d、40 d、50 d、60 d 浸泡时间下露酒原液中的挥发性有机物定性分析见图20、图21、图22、图23、图24、图25、图26 和图27;不同浸泡时间的露酒气相离子迁移谱图定性结果见表2。

图20 浸泡0 d露酒原液挥发性物质定性分析图

图22 浸泡10 d露酒原液挥发性物质定性分析图

图23 浸泡20 d露酒原液挥发性物质定性分析图

图24 浸泡30 d露酒原液挥发性物质定性分析图

图25 浸泡40 d露酒原液挥发性物质定性分析图

图26 浸泡50 d露酒原液挥发性物质定性分析图

图27 浸泡60 d露酒原液挥发性物质定性分析图
从图20—图27并结合表2分析发现,该复合型露酒原液的主要挥发性成分为酯类和醇类物质,其中部分物质在桂花香气挥发性物质中也有检出[40],但是大部分醇类物质为浸泡绵柔型基酒所带入,它们对于露酒的香气挥发具有携带作用,使得露酒各种风味物质之间协调。露酒随着浸泡时间不同其风味物质逐渐发生变化,但是并不是随着时间的延长,其风味成分呈现简单的线性关系,而是呈动态的变化趋势,这可能与露酒浸泡过程中各种生物活性成分的变化有关(结合、分解等),同时该复合型露酒原液的浸泡是在室温的条件下进行的,可能后期随着浸泡时间的延长,与温度的变化(室温逐渐变低)也有一定的关系,如该浸泡条件下的0、5 d、20 d、40 d、60 d 呈现一定规律,10 d、30 d、50 d相似。
FlavourSpec®风味分析技术,对于产品的特征图谱建立有着重要的意义,尤其是在该露酒成品后期的追踪溯源技术上,有着重要的地位。甚至可以区分不同产地、原料、年限等露酒的风味物质等,建立不同露酒挥发性有机物的数据库,成为露酒指纹图谱的推动者,进一步建立行业标准。
3 小结
本研究主要探讨了复合露酒浸泡过程中黄酮、多酚、皂甙、总糖等活性成分、色度及挥发性成分的变化情况,结果发现复合露酒浸泡过程色度变化源于黄酮、多酚、皂甙等物质的浸出情况,随着这些活性物质浸出量增多,L 值下降,a*和b*值上升。通过实验发现,浸泡物料浓度为5 %(m/v)、基酒酒精度为45 %vol,浸泡时间30 d,此时复合露酒中黄酮、多酚、皂甙、总糖含量分别为0.1335 mg/mL、0.0932 mg/mL、1.8707 mg/mL 和0.0833 mg/mL,色度中L,a,b 值分别为:66.7983、17.7183 和81.4383,此外超声处理时间50 s,间隔时间60 s,超声次数20~30次,可使复合露酒的黄酮等活性成分及色度快速达到平衡。随着复合露酒浸泡时间的延长,其挥发性成分也发生一定的变化,其中挥发性有机物的含量随浸泡时间延长变化较小的主要有乙醇、丁酸乙酯、2-甲基-1-丙醇、3-甲基丁醇和丁醇等;随着浸泡时间延长挥发性物质显著下降的主要有乙酸异戊酯和2-丁酮等;部分挥发性有机物的含量随浸泡时间延长逐渐升高,主要有己醇、乙酸乙酯、己酸乙酯、2,6-二甲基吡嗪、戊酸乙酯、2-甲基丁酸乙酯和3-甲基丁酸乙酯等。这些挥发性有机物的变化消长促使复合露酒的风味趋于成熟。
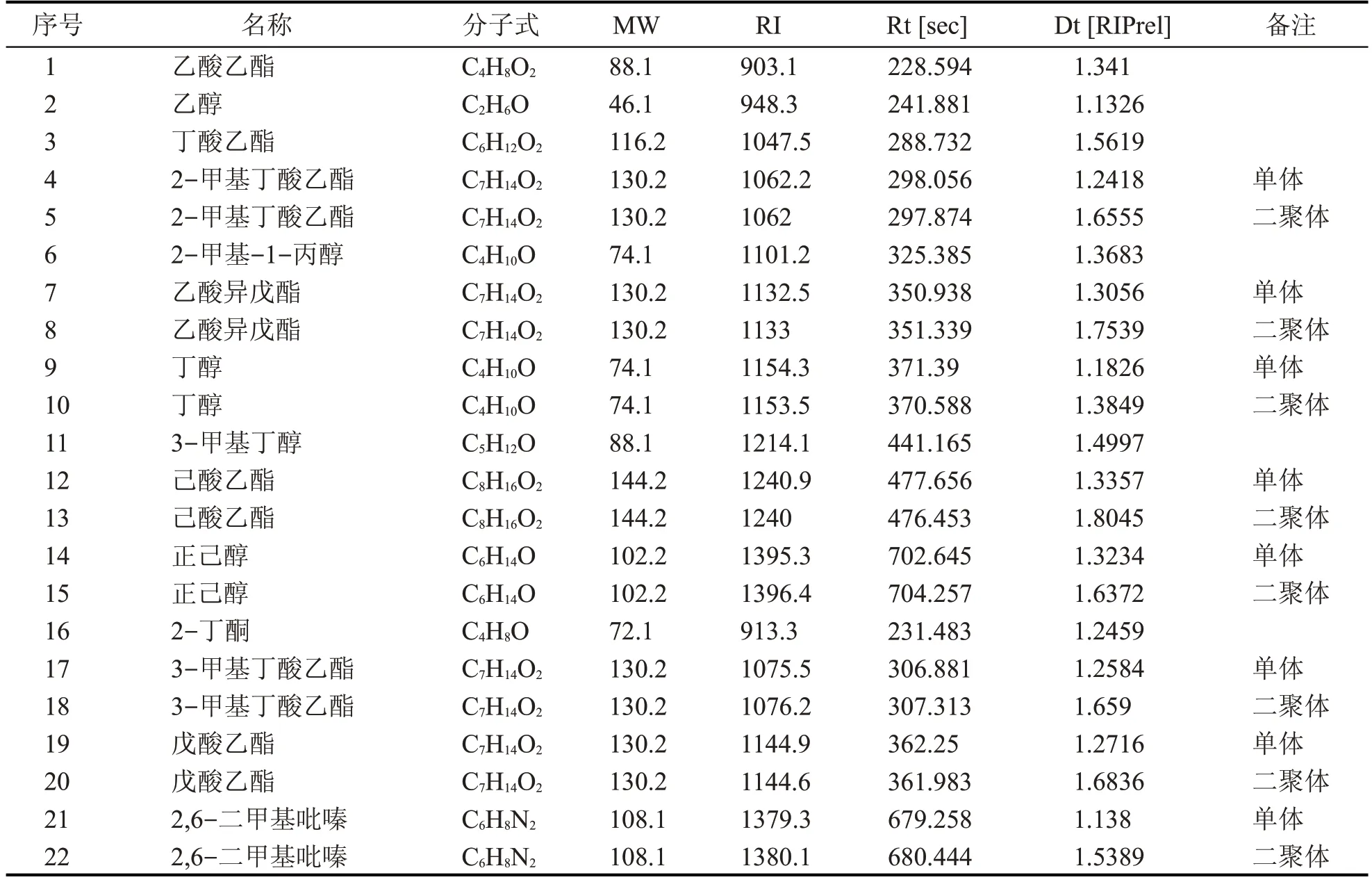
表2 不同浸泡时间下露酒的气相离子迁移谱图定性结果
