Akt2-Bad信号通路参与NSCLC凋亡*
2021-04-01蒋莉何芳刘丹李为民陈渤江
蒋莉 何芳 刘丹 李为民 陈渤江
(1.川北医学院附属医院呼吸与危重症医学科,四川 南充 637000; 2.南充市中心医院·川北医学院第二临床医学院,四川 南充 637000;3.川北医学院,四川 南充 637000;4.四川大学华西医院呼吸与危重症医学科,四川 成都 610041)
磷脂酰肌醇3激酶(PI3K)/Akt/mTOR信号通路是细胞内主要的信号通路之一,在细胞内基本功能中起着重要作用。 PI3K/Akt/mTOR通路调节细胞增殖、生长、细胞大小、代谢和运动。这一通路已被广泛研究,并被证实在人类肿瘤中普遍被激活。Bad 既是Bcl-2家族的一员,也是PI3K/Akt信号通路下游的调节因子。PI3K活化可激活PI3K依赖性的Akt,减少Bad与Bcl-XL和Bcl-2结合,阻止细胞色素C释放入胞浆,而抑制细胞凋亡[1-2]。EGFR/MEK/MAPK通路激活p90Rsk通过磷酸化Bad,调节细胞增殖、分化和凋亡[3-4]。除此之外,很多细胞因子都可通过使Bad失活而抗凋亡[5]。譬如:IL-3、PKA都可以通过细胞内信号转导通路使Bad失活,从而抑制Bad的促凋亡活性[6]。由此可见,Bad的调节是由多个蛋白之间相互作用而形成的复杂网络来实现的。
我们先前的研究发现,Bad过表达可抑制非小细胞肺癌(Non-small cell lung cancer,NSCLC)的增殖,促进NSCLC尤其是肺腺癌细胞株H1299、SPC-A1、H292及大细胞肺癌H460的凋亡[7]。靶向沉默Akt2的表达也可以抑制NSCLC细胞增殖,促进其凋亡[8]。然而在NSCLC中,是否通过Akt2/Bad信号通路的形式来实现对肿瘤细胞凋亡的影响,目前尚不清楚。因此,我们构建了Bad过表达同时稳定干扰Akt2的NSCLC细胞株,通过检测体外细胞凋亡及凋亡信号通路下游蛋白,并选择肺腺癌细胞株H1299、SPC-A1构建移植瘤模型,探讨Akt2/Bad信号通路在NSCLC中的作用机制。
1 材料与方法
1.1 材料 人肺腺癌细胞株SPC-A1、肺癌细胞株(淋巴结转移)NCI-H292、人高转移大细胞肺癌细胞株NCI-H460、人肺腺癌细胞株NCI-H1299、肺鳞癌细胞株SK-MES-1,以上细胞株购于中国科学院典型培养物保藏委员会细胞库;慢病毒载体及包装系统载体pLOV.UBC. EGFP载体、膜蛋白外壳质粒和反式包装质粒三种质粒购于纽恩(上海)生物科技有限公司;Enhanced infection solution(ENI.S)、polybrene、含无义序列的shRNA及绿色荧光蛋白(GFP)标记对照慢病毒载体、小干扰序列和引物购于上海吉凯基因技术公司;2000DMEM、FBS购于hyclone;primer(R&F)、Trizol、Lipofectamine、positive clone 测序及逆转录试剂盒购于Invitrogen公司;BamHI、EcoRI等限制性内切酶购于New England Biolabs;SYBR TAQ Real-time试剂盒购于Bio-Rad公司;质粒抽提试剂盒购于Qiagen公司;Cell Counting kit-8 (CCK-8)购于Dojindo公司;V-APC/7AAD、蛋白抽提试剂盒购于南京凯基生物科技公司,BCA protein assay kit购于Pierce公司;兔抗人AKT2抗体、抗兔二抗购于 Cell Signaling Technology;Luminata Western HRP substrate、PVDF膜购于Millipore公司;预染蛋白Marker购于Fermentas公司。
1.2 制备实验所用NSCLC细胞株 制备稳定干扰Akt2表达的同时稳定过表达Bad基因的NSCLC细胞株(Akt2+Bad),具体方法详见课题组之前的研究[7-9]。
1.3 实验分组 未感染慢病毒载体的H1299、H292、SPC-A1、H460及SK-MES-1细胞为NSCLC组。阴性对照病毒转染NSCLC细胞为NC组; shAkt2组:稳定干扰Akt2的NSCLC细胞;Bad组:稳定过表达Bad的NSCLC细胞;shAkt2+Bad组:稳定干扰Akt2表达同时稳定过表达Bad基因的NSCLC株。
1.4 Western blot检测蛋白水平 根据凯基生物蛋白抽提试剂盒操作说明抽提细胞蛋白。经15%聚丙烯酰胺凝胶电泳,120 V恒定电压转膜,封闭过夜,Akt2一抗、Bad一抗工作浓度1∶1000,二抗工作浓度1∶5000,β-actin一抗工作浓度1∶10 000,二抗工作浓度1∶10 000。采用Millipore公司Luminata Crescendo Western HRP substrate显色液孵育3 min,放入暗盒(感光时间根据荧光条带的强弱来判定)曝光,取出X光片,放入自动洗片机显影定影后用柯达扫描仪将条带扫描至电脑保存。Quantity one分析软件测定条带灰度。
1.5 Annexin V-FITC/PI双染法检测细胞凋亡 用不含EDTA的胰酶消化收集各组细胞。用PBS洗涤细胞2次(2000 rpm离心5 min)收集1~5×105细胞。加入500 μL的Binding Buffer重悬细胞。加入5 μL Annexin V-APC混匀后,加入5 μL 7-AAD染液,混匀。室温、避光、反应5~15 min。在1 h内进行流式细胞仪的观察和检测。激发波长633 nm,最大发射波长660 nm,Annexin V-APC的红色荧光使用FL4通道检测;激发波长Ex=546 nm;发射波长Em=647 nm,7-AAD红色荧光使用FL3通道检测。
1.6 细胞成瘤能力检测 准备雌雄各半、5~8周及体重匹配裸鼠18只,每组6只,SPF级环境饲养。各组细胞常规培养,胰酶消化收集细胞,PBS洗涤两次,PBS重悬。调整细胞浓度,每100 μL约含1×106细胞。无菌操作下,按2×106细胞/只比例,将已制备的细胞悬液经皮下注射于裸鼠的右侧背部,观察细胞成瘤能力。4周后,摘除瘤体,称重、采集照片,将每个瘤体组织分成两部分,一部分10%福尔马林固定,石蜡包埋,另一部分-80℃冻存。
1.7 Tunel细胞凋亡检测 使用红色荧光素(Tetramethylrhodamine,TRITC)标记凋亡细胞中断裂DNA的3-OH末端,并用荧光显微镜检测。石蜡切片按常规方法脱腊,用二甲苯60 ℃浸洗5 min×2次。梯度乙醇各浸洗1次,每次3 min。1×PBS漂洗3次,每次5 min。0.1%Triton X-100处理8 min。PBS洗两遍。取出100 μL Label Solution用于阴性对照;阳性对照使用1500 U/mL Dnase I于15~25℃处理10 min,使DNA断裂。每个样本加入50 μL TUNEL reaction mixture,阴性对照加入50 μL label solution。置于湿盒中防止干燥,37°C避光孵育60 min。PBS洗3遍。50 μL DAPI复染核10 min,PBS洗3遍。抗荧光淬灭剂联合中性树脂封片。10 min后荧光显微镜检测:激发波长543 nm,发射波长571 nm(红)。DAPI最大吸收波长为358 nm,最大发射波长为461 nm。Image pro plus 6.0软件计数细胞。

2 结果
2.1 各组Bad、Akt2的蛋白水平的比较 shAkt2+Bad组Bad蛋白水平表达显著高于NSCLC组和NC组(均P<0.05)。Akt2蛋白水平表达显著低于NSCLC细胞组和NC组(均P<0.05),见图1。
2.2 对体外体内细胞凋亡和细胞成瘤能力的影响
2.2.1 体内实验 shAkt2+Bad组组织切片TUNEL染色与NSCLC、NC、Bad、shAkt2组相比,凋亡细胞数明显增加(P<0.05),这与体外Annexin V/7-AAD流式分析结果一致,见图2A、图3。提示沉默Akt2基因表达与过表达Bad在促进肺癌细胞凋亡方面具有协同作用。
2.2.2 体外实验 shAkt2+Bad组的细胞凋亡率明显高于NSCLC、NC、shAkt2和Bad组细胞(P<0.05),说明靶向沉默Akt2、Bad过表达在对NSCLC细胞凋亡的影响方面存在协同作用。而NSCLC与NC 组凋亡率相比差异无统计学意义(P>0.05),见图2B。说明慢病毒载体本身不会影响NSCLC的凋亡。
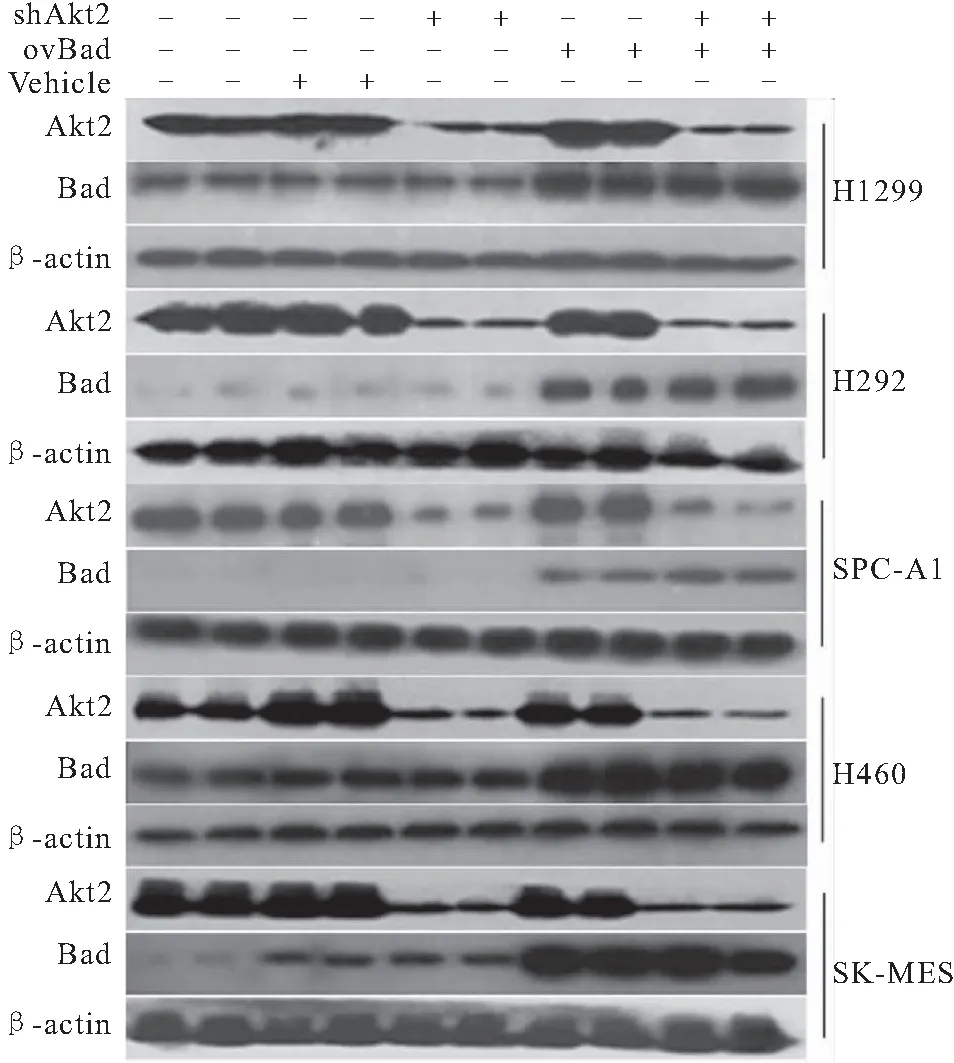
图1 Western blot分析3组细胞Bad蛋白及AKT2蛋白表达水平
2.2.3 细胞成瘤实验 4周后shAkt2+Bad组移植瘤重量较NSCLC、NC、Bad及shAkt2组明显降低(P<0.05),而NSCLC及NC组重量比较差异无统计学意义(P>0.05),见图2C、D。说明靶向沉默Akt2、Bad过表达在对抑制NSCLC细胞成瘤能力的影响存在协同作用,且慢病毒载体本身不会影响NSCLC的成瘤能力。
2.3 对凋亡相关蛋白的调节机制 shAkt2+Bad组瘤体组织中shAkt2蛋白表达水平明显下降,Bad蛋白表达水平明显升高(图4A),提示转染双病毒的NSCLC细胞构建的瘤体组织中, Akt2蛋白稳定沉默且Bad蛋白稳定过表达。与NSCLC及NC组比较,shAkt2细胞株中Caspase-9蛋白表达显著下调,cyto-c显著升高(均P<0.05),而BCL-2和BCL-XL蛋白表达水平均无明显差异(P>0.05);与NSCLC及NC组比较,Bad组细胞株中cyto-c显著升高(P<0.05),而Caspase-9、BCL-2和BCL-XL蛋白表达水平比较差异均无统计学意义(P>0.05);与NSCLC及NC组比较,Bad+shAkt2组中Caspase-9蛋白表达显著下调,cyto-c显著升高(均P<0.05),且cyto-c的增高程度强于Bad和shAkt2组(P<0.05)(图4B)。
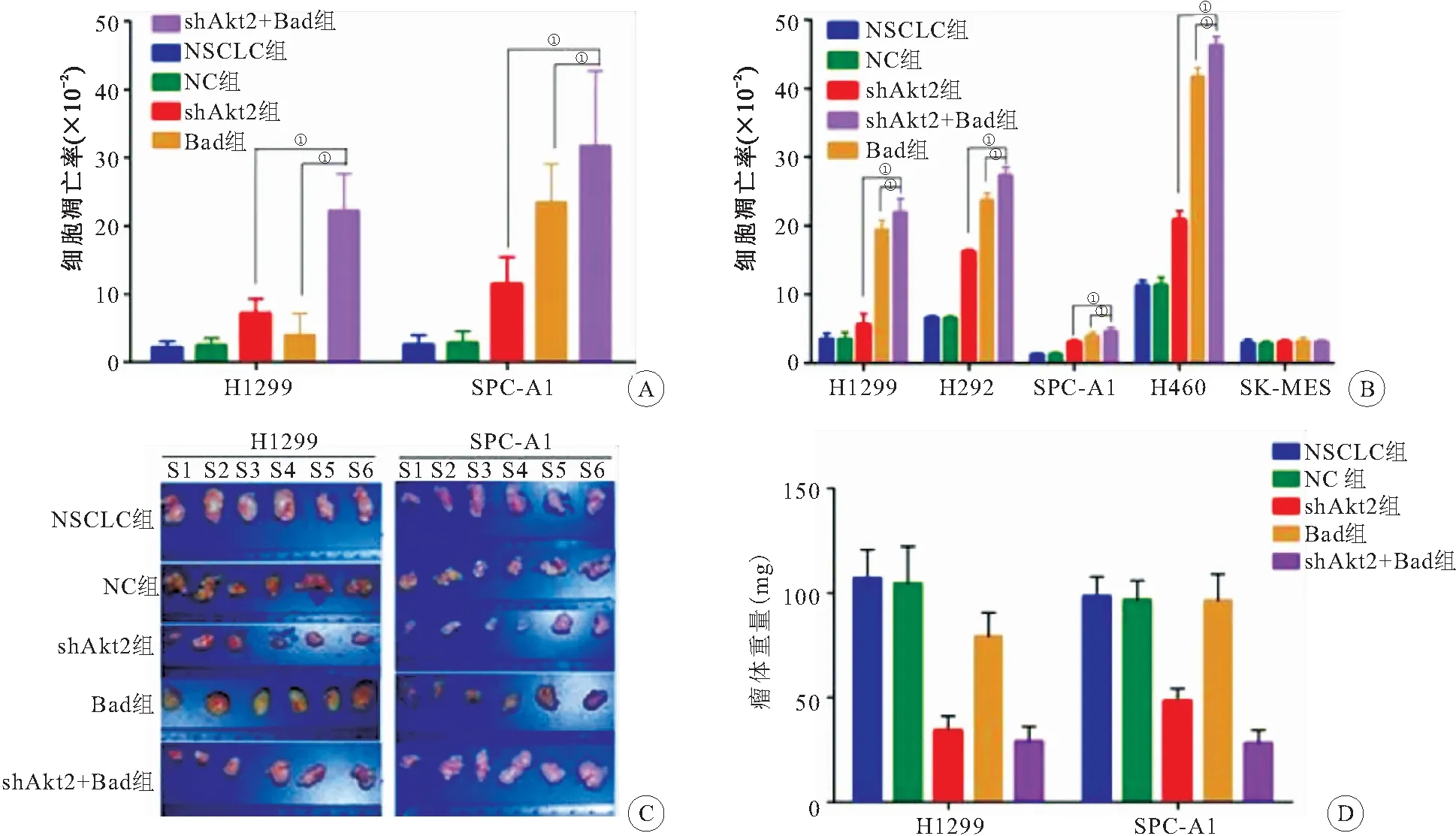
图2 靶向沉默Akt2、Bad过表达对体外体内细胞凋亡和细胞成瘤能力的影响
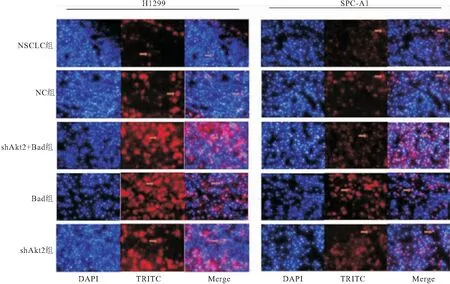
图3 各细胞组移植瘤组织内的细胞凋亡水平(×200)
3 讨论
Akt/Bad信号通路在肿瘤和非肿瘤领域的作用逐渐得到证实:Zhang等[10]发现黄杨酮能抑制Akt/p70s6k1通路,下调Bcl-2家族抗凋亡蛋白表达,上调促凋亡蛋白Bad表达,使Caspase3裂解活化,细胞色素C从线粒体释放到细胞质,从而促进凋亡;在非肿瘤领域, Quan等[11]在弓形虫感染模型中发现,弓形虫感染激活PI3K-PKB/Akt,使Bad磷酸化,抑制宿主细胞凋亡,而用PI3K抑制剂LY294002 和渥曼青霉素能阻止寄生虫诱导的PKB/Akt磷酸化,促进宿主细胞凋亡。李爽等[12]研究发现藻蓝蛋白能够通过细胞周期S期阻滞,调控细胞周期,凋亡和迁移相关基因的表达而抑制非小细胞肺癌H1299细胞的生长和迁移,进而诱导细胞凋亡。但Akt/Bad信号通路是否参与,以何种形式参与NSCLC的发病目前还鲜有报道。
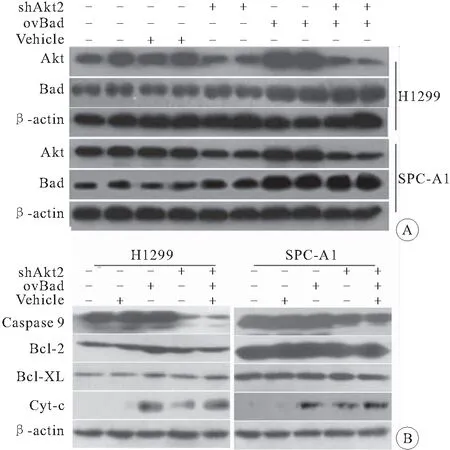
图4 Western blot检测下游分子
细胞色素c(cyto-c)是线粒体呼吸链的重要组成部分,研究表明细胞凋亡的一个关键步骤是细胞色素C 从线粒体释放。释放到细胞质中的cyto-c 在dATP 存在的条件下能与凋亡蛋白酶活化因子1(apoptosis protease activating factor 1,Apaf-1) 结合形成多聚体,并使含半胱氨酸的天冬氨酸蛋白水解酶-9(cysteinyl aspartate specific proteinase,caspase-9)与其结合形成凋亡小体,caspase-9被激活,并激活其他caspase如caspase-3等,促进细胞凋亡[13-14]。本研究体内、体外实验发现,靶向沉默Akt2联合Bad过表达,较单一过表达Bad或敲出Akt2细胞凋亡率明显增加,提示Akt2/Bad信号通路可能以促进凋亡的形式参与NSCLC发病机制。在对凋亡信号通路相关蛋白表达的检测中,本研究结果显示,Akt2靶向沉默和Bad过表达的NSCLC细胞cyto-c显著升高,共感染的NSCLC cyto-c也显著升高,且cyto-c升高的程度大于单病毒感染组。因此,我们的研究提示Akt2/Bad信号通路可能通过线粒体途径调节NSCLC细胞凋亡。但与公认的线粒体促凋亡途径可能矛盾的是,我们的结果显示,靶向沉默Akt2能使caspase-9明显减少。我们采用的caspase-9抗体仅检测细胞总caspase-9含量,有必要在后续的研究中做相关的活性鉴定。
通常情况下,Bad与14-3-3蛋白结合,以磷酸化的失活形式存在于细胞质中,抑制细胞凋亡[15]。外来凋亡信号刺激激活Bad上游信号蛋白,诱导Bad去磷酸化,与BCL-2和BCL-XL结合形成异源二聚体,从而促进细胞凋亡[16]。我们在对凋亡信号通路相关蛋白的检测中发现,沉默Akt2及Bad过表达后,虽然能促进NSCLC凋亡,但BCL-2和BCL-XL的含量均无明显变化,提示调节Akt2及Bad可能只是影响BCL-2和BCL-XL的聚合及解聚而不会改变上述蛋白的总量,或者可能在NSCLC中,凋亡调节并不主要通过上述途径。另外我们发现,靶向抑制Akt2也不会改变其下游蛋白Bad的含量,提示Akt并不依靠改变Bad剂量来调控它的活性。
Chorner等[17]研究发现Akt-1抑制剂a-674563相对于泛Akt抑制剂mk-2206,在降低NSCLC细胞存活率方面更有效。Akt1的缺失可抑制肺癌的发展,而Akt2的缺失则促进了肺癌的发展。p-Akt2是Akt2磷酸化的活性形式。但本研究组既往的研究[18]发现, p-Akt2阳性患者5年生存率明显降低,而p-Akt1与预后无关,推测在NSCLC肿瘤进展中,Akt2发挥更重要的作用。本次研究体内、体外实验再次证实,靶向沉默Akt2能够促进NSCLC的凋亡,抑制NSCLC细胞株在裸鼠体内成瘤。
研究[19]发现,miR-137过表达抑制了顺铂作用下的A549和 H520细胞的Akt2蛋白的表达,增加了caspase-3活性,增加了bax 蛋白的表达,并抑制了cyclin d1蛋白的表达。Akt2抑制剂mk2206可抑制akt2蛋白的表达,抑制顺铂诱导的A549和H520细胞增殖。Akt2的抑制还可以增加caspase-3活性和bax蛋白的表达,该研究结果间接证实了Akt2信号通路在肺癌细胞增殖和凋亡中的作用,并提示这一发现可以转化为抗癌耐药机制的研究。
本研究提示,Akt2/Bad信号通路可能通过线粒体途径参与NSCLC的凋亡,参与NSCLC的发病机制,此外还有多种蛋白构成相当复杂的信号网络参与其中。今后我们将会聚焦在这些网络中寻找关键环节及调控点,并将其转化为抗癌靶向药物的研发和抗癌药物耐药机制的研究。
4 结论
靶向沉默Akt2联合Bad过表达在促进NSCLC细胞凋亡方面有协同作用,能显著增加cyto-c表达,下调caspase-9的表达;Akt2/Bad信号通路可能通过线粒体途径促进细胞凋亡参与NSCLC的发病机制。
