金属基材料电催化CO2还原的研究进展
2021-03-30苏文礼范煜
苏文礼,范煜
(中国石油大学(北京)重质油国家重点实验室,北京102249)
现代工业化社会主要以化石燃料作为能量来源,化石燃料的燃烧会造成大量CO2的排放,而作为温室气体的主要成分,大气中CO2含量的上升会引发全球气候变暖,进而导致生物多样性锐减等严峻的环境问题。同时,随着社会的高速发展,传统的化石能源逐渐枯竭,使得人们开始致力于发展新型可再生能源。利用CO2作为生产各种燃料和化学物质的来源,是减少碳排放和保障能源供应的有效策略之一。目前,转化CO2的方法主要有化学热还原、光催化、电催化和生物还原等。其中,电催化还原CO2具有易于直接控制、以可再生的电能驱动、能将CO2转化为多种碳产物、通常在室温常压下进行等优点,受到广泛的研究。虽然其具有很大的潜力,但由于缺乏高效的电催化剂,目前电化学还原CO2的能量转化和化学转化效率依然受到限制。因此,研制高活性、高选择性并且能够在较低的过电位下还原CO2的电催化剂成为研究人员奋斗的目标。在众多催化剂中,金属基材料由于其高导电性,在电化学反应中表现出较好的活性,并且具有空轨道和活跃的d 电子,对CO2有较强的吸附和脱附能力,被广泛应用于电催化CO2还原。本文将主要介绍水溶液介质中金属基材料电催化CO2还原及其反应途径的研究现状。
1 电催化CO2还原概述
CO2是有机物燃烧的最终产物,化学状态相当稳定,因此活化CO2分子的能垒很高,需要较高的过电位才能促使电催化反应发生[1]。另一方面,电化学还原CO2反应涉及多电子传递机理,不同反应条件下可以生成一氧化碳、甲酸盐、甲醇、乙烯、乙醇等多种产物[2-3]。根据热力学研究,对于不同的产物,还原半反应的平衡电位相近[见表1,其中电极电位以相对于标准氢电极(standard hydrogen electrode,SHE) 表示,在25℃、1atm(1atm=1.01325kPa)和pH=7 的标准试验条件下测得];同时,由于在水溶液中具有与CO2电化学还原反应相当的热力学电势,析氢反应(HER)也是一个重要的竞争反应[4]。动力学和热力学两方面的特点表明,缓慢的化学反应速率和伴随的副反应是有效还原CO2为特定产物的主要障碍。影响电化学还原CO2反应的因素主要包括电极电势[5]、电催化材料[6-7]、电解质溶液[8]和压力温度[9-10]等,其中电催化材料对还原反应的产物分布和选择性起着决定性的作用。早在20世纪80年代,Frese 等[11]就将数种金属和半导体作为电极材料用于还原CO2,Hori等[12]发现Cu 金属能将CO2电化学还原为CH4和C2H4。迄今为止,绝大多数金属都被用于电化学还原CO2的研究[4]。Hori等[13]在前期工作中根据反应产物的不同,将金属电催化剂大致分为4类:①金属In、Sn、Hg 和Pb 等主要生成HCOO-;②金属Au、Ag 和Zn 等主要生成CO;③金属Fe、Pt、Ni 和Ti等由于析氢电位过低,几乎只生成副产物氢气;④金属Cu 表现出独特的催化特性,能以可观的选择性生成多种烃类和醇类。但是这些传统的多晶单金属催化剂在反应中往往过电位较高,选择性不佳,对竞争反应(HER)的抑制效果也较弱。因此,开发出高效稳定的电催化材料成为CO2电化学还原技术工业化应用的关键。

表1 电化学还原CO2半反应的电极电位(标准试验条件)[4]
2 反应机理
电催化CO2还原反应机理的复杂性给高效催化材料的开发带来了很大的困难。为了解CO2在不同催化剂表面上还原的途径,进而指导催化剂的开发,研究人员进行了大量的实验和理论计算,目前已取得了一些成果。
最早Hori 课题组[5]和Bard 课题组[14]根据实验现象推测了CO2在铜电极上还原的反应途径,而后密度泛函理论(density functional theory,DFT)等理论计算工具的高度发展极大促进了CO2电化学还原反应的机理研究,这些理论计算可以提供反应中间体与催化剂表面的结合能、基元反应的自由能和活化能垒等信息,进而评估不同反应途径的可行性和确定有利于反应的关键中间体[15-16]。与此同时,先进的原位分析设备可以直接观察反应过程中的表面结合物以及其与催化剂的相互作用,为基于理论计算提出的反应机理提供了重要的证据[17-18]。

图1 电催化还原CO2为C1产物的反应途径
当前,CO2在水溶液中电化学还原为HCOO-和CO的反应路径已经比较清晰(图1[19])。Cui等[20]提出在生成HCOO-的过程中,羟基化的SnO表面会首先形成碳酸氢盐,然后进一步还原为*OCHO或*COOH。与Sn、In 不同,还原反应中在Pb 催化剂表面没有观察到碳酸氢盐中间产物的形成,因此Pander等[21]认为CO2在Pb表面上还原是通过与被吸附的*H 反应进行的。并且,许多研究都表明在生成甲酸盐的过程中,*OCHO 是一种比*COOH 更有效的中间体[22-24]。而与甲酸盐的生成机理不同,研究者一般认为电化学还原CO2为CO 是通过中间产物*COOH 完成的[图1(b)][25]。Firet 等[26]在反应过程中利用原位衰减全反射傅里叶变换红外光谱(ATR-FTIR),直接证明了*COOH 的存在,根据实验结果,认为*COOH 的形成可能通过两种途径:①在中等过电位下,CO2通过质子耦合电子转移形成;②在更高的过电位下,存在首先通过单电子转移形成*COO-,然后再经质子化生成*COOH 的步骤。吸附的*COOH 继续接受电子和质子,随着水分子的脱除进一步被还原为*CO,最后*CO 脱附生成CO[27]。基于以上的反应机理,电化学还原CO2为CO 的有效催化剂应当具有较强的*COOH 结合能和较弱的*CO 结合能。然而,Hansen 等[28]的DFT 计算结果显示,中间产物*CO和*COOH与金属催化剂表面的结合能具有很强的比例关系,不可能在同一活性位点增强*COOH吸附强度的同时降低*CO的吸附强度。因此,优化催化剂与中间产物*COOH 的结合能成为提高其CO选择性的关键。
目前为止,能将CO2电化学还原为烃类和醇类的有效催化剂仍然绝大部分是铜基材料,但与生成HCOO-和CO 的反应机理相比,在Cu 电极上将CO2还原为多碳化合物的反应路径更为复杂。经过多年探索,研究人员普遍认为*CO是生成C2产物的关键中间体,而金属铜独特的催化特性正是来源于其与*CO 适宜的结合能[25,29-30],但是*CO 进一步还原为C2产物的具体步骤目前尚存在争议。Garza 等[31]基于实验发现和DFT 计算,提出了一种铜电极上电化学还原CO2为C2产物的反应途径[乙烯和乙醇的生成途径,图2(a)],研究认为催化剂表面吸附的*CO 会首先加氢形成*CHO,再与游离的CO 分子耦合形成*COCHO,而*COCHO 作为生成C2H4和C2H5OH的关键中间产物,会通过形成*COCHOH 进而生成C2H4(乙烯路径),或者生成CHOCHO后再与催化剂结合形成CH2CHO*中间产物,最后生成C2H5OH(乙醇路径);该研究的计算结果表明乙烯路径和乙醇路径在U=-1.0V时的能垒分别为0.49eV和0.58eV,乙醇路径更高的能垒也解释了一般铜基催化剂对乙烯选择性更佳的原因。而Goddard 等[32]则认为,首先形成的C2中间体为*COCOH,然后形成关键中间产物*CHCOH,经*CHCOH→*CHCHOH路径生成C2H5OH,或经*CHCOH→*CCH 路径生成C2H4[图2(b)]。此外,近年来有研究者将Cu 基双金属催化剂用于CO2电化学还原反应时发现其对C2H5OH 的选择性超过了C2H4,经过分析提出了一种生成C2H5OH 的插入机理,对CO 吸附较弱的Zn或Ag 等位点上产生游离的CO,然后迁移插入Cu表面与*CH2之间形成中间产物*COCH2,最后还原为C2H5OH[33-34]。当前的电催化剂对C2H6的选择性普遍不高,Dutta 等[35]认为C2H6是通过C2H4进一步加氢还原生成的,因此需要额外的能量。而Handoko等[36]则提出在低过电位下,C2H6也可能通过中间体*CH3二聚形成。由前文可见,尽管近几年来电化学还原CO2的机理研究成果显著,但由于实际反应的复杂性,该领域对于C2产物的形成途径还未达成共识,主要的争议是*CO 二聚耦合途径是经过*COCHO 还是*COCOH,以及乙烯与乙醇路径发生偏离时的关键中间体。并且,目前在构建反应路径的理论计算中普遍忽略了动力学因素、溶剂效应和电势等的影响,为充分理解在真实反应条件下的机理,需要建立更精确的模型来描述复杂的CO2电化学还原过程。原位计算模型结合原位光谱和电化学实验,在原子/分子水平上理解电化学还原CO2的反应机理有助于开发出更为高效和稳定的电催化剂。
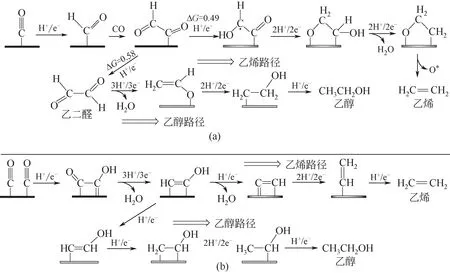
图2 铜电极上电化学还原CO2为C2H4和C2H5OH的反应途径
3 电极材料
Kibria等[37]将电化学还原CO2与电解水类比,利用技术经济分析认为,电流密度要达到300mA/cm2,法拉第效率(Faradaic efficiency,FE)达到80%~90%,电压低于1.8V,并且能够保持80000h 以上的稳定性,电催化CO2还原的工业化应用才具有可行性。但是传统的多晶单金属材料在CO2电化学还原反应中表现不佳,为提高催化性能,研究者采用纳米化[38-39]、合金化[40]、氧化处理[41]以及与其他材料复合[42]等手段对传统多晶电极进行改性,显著增强了金属电极的活性和选择性,这些新型催化剂代表了CO2电化学还原的新趋势。最近几年来,达到原子级分散的单原子催化剂[43-44]也被应用于CO2电化学还原,由于其独特的几何和电子结构,在反应中显示出优异的催化性能。为便于读者快速查找关键信息和更直观地比较,本节首先对后文讨论中涉及的各类型催化剂的性能进行了总结[表2,其中电位以相对于可逆氢电极(reversible hydrogen electrode,RHE)表示,下同]。
3.1 金属纳米催化剂
相比于传统块状金属,纳米结构的金属电极一方面提高了比表面积,增加了催化活性位点的数量;另一方面,纳米催化剂表面具有大量配位不饱和的位点,可以显著提高催化活性,改善电催化性能。金属纳米催化剂的尺寸、结构和形貌对CO2电化学还原反应的效率和产物分布都具有非常重要的影响。Reske 等[72]制备了一系列不同尺寸的铜球形纳米颗粒用于电催化CO2还原,电化学实验表明:随着颗粒尺寸的降低,特别是低于5nm 时,H2和CO 的选择性上升,而CH4和C2H4的选择性降低;分析认为纳米颗粒的尺寸减小后,表面上低配位数原子所占的比例升高,这些低配位数的表面原子具有更强的结合能,使得中间产物*CO和*H的迁移率降低,不利于CO 的后续加氢,所以CO 和H2的选择性升高,CH4和C2H4的选择性降低。Loiudice等[45]制备的更大尺寸(44nm)的铜立方体纳米颗粒(Cu NCs)则对C2H4的选择性更高,在电位为-1.1V 时,C2H4的FE 达到41%。Jung 等[46]发现在电化学还原CO2的反应过程中,20nm 的立方体形Cu2O纳米颗粒逐渐破碎为2~4nm的小颗粒,与此同时,产物C2H4的FE 从27%逐渐上升到57.3%,并且更为有效地抑制了析氢反应;分析认为,这种罕见的催化性能提升与催化剂破碎过程中铜的化学状态密切相关,破碎后小纳米晶之间的紧凑分布也有利于C2H4的生成。此外,Mistry 等[73]认为反应过程中反应物在颗粒之间的扩散和中间产物的再吸附等介观现象同样不可忽视,因此纳米颗粒之间的距离也是影响产物分布和选择性的重要因素。Jeong等[47]进一步地调控了Cu 颗粒中两个晶面之间的原子尺度间距(ds),电化学实验表明,与未经优化的CuOx纳米颗粒相比,ds为0.5~0.6nm 的Cu 颗粒电流密度提高了近12倍,并且C2+产物的FE达到了80%。
除了金属纳米颗粒外,暴露更多催化活性位点的一维、二维和三维纳米电极也被用于电催化CO2还原。Zhu 等[48]合成了一系列宽度约为2nm、长度不同的超薄Au 纳米线(Au NWs)作为还原CO2的电催化剂,电化学实验表明:与块状Au电极相比,Au NWs 可以显著降低起始电位,并且生成CO 的选择性最高可以达到94%,持续反应6h 后活性没有明显降低;通过DFT计算认为Au NWs优异的催化性能来源于高密度存在的边缘位点,这些位点有利于CO2活化为*COOH,并且可以促进*CO的脱附。受此启发,Li等[49]在160℃下还原油胺中的CuCl2和三甲基硅烷,合成了宽度约为20nm 的铜纳米线(Cu NWs),由于高密度边缘位点的存在,Cu NWs在电位为-1.25V时电催化CO2还原为CH4的FE达到了55%[49]。Luc 等[50]合成了一种独立式三角形形状的二维Cu 纳米片,能够选择性地暴露Cu(111)晶面;在CO电化学还原反应中,该Cu纳米片对C2+产物的总FE 达到了70%,其中乙酸盐的FE 最高为48%,并且乙酸盐的部分电流密度高达131mA/cm2,稳定性测试表明持续反应3h后乙酸盐的FE没有明显降低。Xie 等[51]则通过原位电化学脉冲氧化铜箔制备出了一种形貌独特的三维菊花状Cu 纳米材料(Cu NFs),可 以 催 化CO2生 成HCOOH、CH4和C2H4,在较宽的电位范围内能够明显抑制析氢副反应的发生。Hossain 等[74]报道了一种新型的铜纳米枝晶催化剂,独特的树枝状形貌和较大的活性表面积使其在CO2电化学还原反应中表现出优异的催化活性。Luo 等[52]利用电沉积方法制备的三维分级多孔In 催化剂(hp-In)可以将CO2高选择性地还原为HCOOH,电位在-1.0~-1.2V 内,HCOOH 的FE能达到90%左右,其产率最高为1.14mmol/(cm2·h),且能保持24h的长期稳定性,DFT计算的结果表明相比于其他中间体,生成HCOOH 的重要中间产物*OCHO在hp-In催化剂表面更加稳定。并且多孔In还可以作为模板合成多孔Pd-In催化剂,用于调节HCOOH 与CO 的选择性,表现出极大的应用潜力。总之,通过优化金属纳米催化剂的尺寸、结构和形貌,能够显著提高其电化学还原CO2的催化性能,然而纳米电催化剂明确的构效关系当前仍然不清楚,将来还需要更多的研究以加深对影响产物选择性因素的理解,进而指导高效催化剂的设计与开发。
3.2 金属合金催化剂
物质中单个原子的电子特性会受附近原子的影响,在单金属材料中掺杂其他金属元素后,不同元素原子之间的相互作用会显著改变其电子结构,进而改变中间产物在催化剂表面的吸附强度;此外,活性位点原子排列方式的变化也会改变中间产物与表面的吸附方式。因此,将金属合金化也是调整金属电极催化性能的有效手段。Rasul 等[53]将铟电沉积到粗糙的铜表面,制备了Cu-In合金催化剂,用于电催化还原CO2,尽管相比于未沉积In的单金属Cu 电极,合金催化剂的电流密度并没有明显的提高,但合金催化剂抑制了析氢反应的发生,显著提高了CO的选择性,在-0.7V的电位下,CO2转化为CO的FE达到了95%,远高于单金属Cu或In电极,并且在-0.6V的电位下持续反应7h后,CO的FE能够稳定在85%左右;理论计算表明In 和Cu 的相互作用会排斥H 的吸附,从而抑制析氢副反应的发生,而对CO 的吸附能没有明显影响,因此提高了CO2还原反应的选择性。Chen等[54]利用预合成的Cu纳米颗粒与Pd2+前体之间的电偶取代反应,合成了粒径约为2nm 的超细CuPd 纳米合金催化剂,并通过改变Cu/Pd的原子比例来调控其表面组成;电化学实验结果表明:Cu5Pd5纳米合金在电位为-0.87V时,电催化CO2转化为CO的FE约为88%,相应的电流密度约为4.5mA/cm2,在20h 的连续反应中也表现出较好的稳定性。
除了CO,金属合金化的方法也可提高HCOOH的选择性。Ye 等[55]通过改良的电沉积方法制备的Sn-Cu 合金催化剂在电位为-1.14V 时,对HCOOH的FE 约为82.3%,并且,HCOOH 的部分电流密度高达1490mA/mg,是目前报道的除贵金属基催化剂外最高的质量活性。Kwon等[56]首先采用溶胶-凝胶法合成In2O3-ZnO 纳米复合材料,然后通过原位电化学还原,制备了不同In/Zn 比例的In-Zn 合金催化剂;CO2电化学还原反应结果表明,In/Zn比例为0.05 的In-Zn 合金催化剂在-1.2V 的电位下,生成HCOOH 的FE 为95%,HCOOH 的 产 量 达 到 了0.4mmol/(cm2·h),以及实现了24h 的稳定反应。铜基合金催化剂对C2+产物也表现出了可观的选择性,Hoang 等[57]在含有3,5-二氨基-1,2,4-三唑(DAT)作为抑制剂的溶液中,通过电沉积制备了CuAg 合金薄膜,这种合金催化剂在CO2电化学反应中对C2H4和C2H5OH的FE最高分别为60%和25%,总电流密度可以达到300mA/cm2,且此时过电位仅-0.7V。Shen 等[58]结合电沉积和原位电化学还原,将AuCu 合金嵌入Cu 亚微锥阵列中制备的催化剂(AuCu/Cu-SCA)在电位为-1.0V时,将CO2转化为C2H5OH 的FE 达到了29%,其部分电流密度约为5.59mA/cm2,同时抑制了C2H4的生成,其FE 仅为16%,部分电流密度为3.09mA/cm2,且持续反应24h后,催化剂的活性和选择性没有明显变化。此外,Kortlever 等[75]还将对中间产物*CO 吸附较强的Pd 和吸附较弱的Au 合金化,通过优化与*CO 的结合能,促使其进一步还原,结果表明Pd-Au 合金催化剂能够将CO2还原为多种C1~C5烃类,尽管其选择性都不高,但也为设计能高效还原CO2为C2+产物的非铜基电催化材料指明了方向。总之,通过改变合金催化剂的组成和结构,不断优化其元素分布方式,能够有效调节催化剂对中间产物的吸附能力,从而引导目标产物的生成。
3.3 氧化物衍生的金属催化剂
预先将金属氧化,然后在反应过程中进行原位电化学还原是另一种对金属电极改性,以提高其在CO2电化学还原反应中催化性能的方法。这种方法首先由Frese 等[76]报道,氧衍生的铜电极提高了甲醇的选择性。最初主要采用在空气中退火的方法将金属预氧化,Li 等[77]通过高氧化温度和长氧化时间,将Cu 箔在空气中退火处理后得到表面为Cu2O层的纳米线形催化剂,该催化材料相比于多晶铜箔,电化学还原CO2的过电位降低了约0.5V,但主要生成CO和HCOOH。该课题组[59]还利用相似的方法合成了氧衍生的金电极,在电位仅为-0.35V时,8h 的持续电解能够维持2~4mA/cm2的电流密度和超过96%的CO 选择性。与退火后还原的方法不同,通过电沉积金属铜氧化物再还原能够促进C2产物的生成[78]。Ren 等[60]通过恒电流沉积的方法在抛光的铜基底上制备了Cu2O 薄膜,用于电化学还原CO2时产物C2H4和C2H5OH 的FE 最高可以达到34%~39%和9%~16%,而CH4的形成明显受到抑制。氧等离子体处理是另一种对金属进行预氧化的方法,并且相比于热氧化,氧等离子体处理可以在室温下快速改变催化剂表面的化学状态,其过程更为简便和可控,尤其是金属铜经氧等离子体处理后能显著提高C2产物的选择性[61-62,79]。Mistry等[61]利用经氧等离子体处理后的铜箔电催化CO2还原,产物C2H4的FE最高可以达到60%。而Gao等[62]利用氧等离子体技术制备的铜基纳米立方体催化剂,对C2和C3产物的总选择性达到了73%。除此之外,在反应过程中对金属电极原位氧化也是一种很有前景的方法,例如Engelbrecht 等[80]将CO2/O2的混合气作为原料气,对金属铜电极进行原位氧化能够明显抑制CH4的生成,提高C2H4的选择性。
尽管氧化物衍生的金属电极在电化学还原CO2的反应中表现出了优异的催化性能,然而其催化机理和活性位点尚存在争议。Mandal 等[81]的研究表明:在电化学反应过程中会首先将氧化态的铜还原为金属铜,因为氧化铜的还原在动力学和能量上都比CO2还原更有利;并且Lum等[82]利用18O同位素探究了残留氧化物的稳定性,结果表明:在CO2电化学反应后最初的18O 仅有很少一部分残留(<1%),因此认为氧化物衍生的金属电极对CO2还原反应优异的催化性能与残留的氧化物关系不大。然而,最近越来越多的研究认为Cu0、Cu+以及它们的界面是氧衍生的铜催化剂的活性位点[83]。根据理论计算,催化剂次表面Cu+和O 的存在能够促进CO2分子的活化以及中间产物*CO 的二聚[84-85]。Mistry 等[61]通过电化学测试和光谱表征结果提出在反应过程中依然有Cu+存在,并且认为Cu+对于降低起始电位和提高C2H4选择性至关重要。此外,很多研究发现通过调整Cu 的氧化状态,可以显著改变CO2电化学还原反应的产物分布[86-87]。基于这些发现,设计能使Cu+在还原反应过程中稳定存在的电催化剂或许可以进一步提高CO2电化学反应中C2+产物的选择性。
3.4 金属基复合催化剂
在电催化领域,金属材料的研究最为广泛,但普通金属电极在CO2还原过程中依然存在电流效率较低、稳定性不佳等缺点。具有高比表面积和电导率的碳材料由于易于修饰和功能化的特点,常被用作催化剂载体。将金属负载在杂原子掺杂的碳材料上,金属与载体的相互作用可以改变金属的电子结构,进而提高其本征催化活性,有利于CO2分子的活化和调整重要中间产物与催化剂的结合能,是一种提高金属电极还原CO2催化性能的高效方法[42]。Song等[63]通过电化学成核的方法,将铜纳米颗粒负载在表面具有大量的褶皱和尖刺且掺杂了氮的多层石墨烯(CNS)上,制备了一种铜基复合材料(Cu/CNS),相比于负载在玻碳电极上的铜纳米颗粒和没有负载铜的CNS 材料,这种Cu/CNS 复合电极在电化学还原CO2反应中展现出了更优异的催化性能:电位为-1.2V 时,C2H5OH 的FE 高达63%,而前两者几乎没有C2H5OH 生成,且电流密度分别为前两者的3 倍和5 倍,持续反应6h 后,Cu/CNS催化剂的活性和选择性没有明显降低;通过电化学分析和DFT 计算,初步认为铜纳米粒子和氮掺杂的碳纳米尖刺之间的协同效应,在中间产物*CO二聚为C2H5OH 的过程中发挥着关键的作用。Zhang等[64]将前体通过热处理得到预成型的In(OH)3与还原氧化石墨烯(rGO)的混合物,然后在高温下退火制备了In2O3-rGO 复合催化剂,由于In2O3与rGO 之间的化学耦联作用,使得rGO能够更大程度地将电子转移给In2O3,提高了催化剂的电导率,有利于界面上电子的转移,并且降低了形成重要中间产物HCOO*的吉布斯自由能;在电位于-0.6V 后,产物HCOOH 和CO 的总选择性能够达到92%左右,而电位为-1.2V时,主要产物HCOOH的FE为84.6%,其部分电流密度约为22mA/cm2,且连续反应10h后,催化剂的活性和选择性没有明显降低。LYU等[65]将银纳米颗粒固定在石墨烯包裹的氮掺杂碳泡沫上制备了Ag-G-NCF 复合催化剂,在CO2电化学还原反应中产物C2H5OH 的FE 高达82.1%~85.2%,且此时电位仅为-0.6~-0.7V,可以稳定反应10h,但电流密度较低,电位为-0.5V 时仅为0.31mA/cm2左右;该研究认为其超高的乙醇选择性是由于吡啶氮能够增加中间产物*CO 在催化剂表面的吸附强度,促使其转化为*OC-COH,最后生成C2H5OH。这为开发能有效还原CO2为C2产物的非铜基电催化材料提供了新方向。
除石墨烯外,生物质碳材料由于其具有较大的电导率、耐腐蚀性较强等优点,近年来也被广泛应用于电化学领域。Huo等[66]利用蝴蝶的翅膀辅助制备了一种含单分散铜颗粒的电催化剂(Cu Ps/BCF),由于吡啶氮与铜颗粒之间的协同效应可以促进C—C 耦合和中间产物加氢,而且蝴蝶翅膀衍生的氮掺杂碳载体可以有效防止铜颗粒团聚,Cu Ps/BCF 电极能以63.7%的FE 将CO2电催化转化为C2H4,且能保持24h的稳定反应。如前所述,金属基复合材料的高催化性能主要来源于金属与载体之间的协同效应,因此如何优化二者的相互作用,以调整中间产物在催化剂表面的吸附强度,是提高金属基复合材料催化活性和产物选择性的关键。
3.5 单原子催化剂
当材料从纳米尺度继续缩小到单个原子级后,其几何和电子结构会发生根本性的变化,因而表现出与纳米材料完全不同的催化特性。这种单原子(SAs)催化剂最早由张涛团队[88]提出。该课题组报道了一种铂原子在氧化铁纳米晶表面上达到原子级分散而不发生团聚的催化材料。由于其优异的催化性能和易于精确调控的特点,单原子催化剂近年来广受关注。Zhao等[67]利用离子交换的方法,将镍的前体分散到以锌为连接位点的ZIF-8表面,然后在高温(1000℃)下加热,由于Zn 的沸点为907℃,因此在加热过程中Zn 节点会蒸发而留下缺陷,这些位点很容易被邻近的镍原子占据,从而形成Ni单原子催化剂(Ni SAs/N-C);在电化学反应中,该Ni SAs/N-C 催化剂将CO2转化为CO 的FE 约为71.9%,电流密度达到10.48mA/cm2,且在60h的持续反应中表现出较好的稳定性。而Pan 等[68]在合成ZIF 时就加入Ni 的前体,Ni2+会部分取代节点位置的Zn2+形成Ni-ZIF,Ni与N之间通过这种化学掺杂的方式能够形成强烈的相互作用,从而使得Ni 在高温处理后几乎完全呈原子级分散,改良制备方法后的Ni-N-C 催化剂在CO2电化学还原反应中也表现出了更优异的催化性能,在电位仅为-0.75V时,产物CO的FE高达97%,此时电流密度为7.51mA/cm2,连续反应10h后电流密度和CO的FE都没有明显降低。Wang 等[69]也采用类似的方法制备了Co 单原子催化剂,并且更进一步地通过改变热解温度精确调控Co 单原子的配位数,实验结果显示:二配位Co-N2单原子催化剂在电位为-0.68V 时,电催化CO2转化为CO 的FE 高达95%,电流密度为18.1mA/cm2,远高于三配位(Co-N3)、四配位(Co-N4)及Co 纳米晶催化剂,且能保持60h 以上的稳定性。此外,Ye 等[89]制备的Fe 单原子材料也在CO2电化学还原反应中表现出优异的催化性能。这种利用金属有机框架(MOFs)材料辅助制备单原子催化剂的方法非常简便而又高效,并且制备的电催化剂在CO2还原反应中表现出了比大多数贵金属基催化剂更高的活性和选择性,极具工业应用潜力。
石墨烯等二维材料具有明确的原子结构和多种化学配位环境,可调节层内受限客体原子的电子特性,也常被用来辅助制备单原子催化剂。Jiang等[70]采用浸渍-还原法得到分散在石墨烯空位中的Ni单原子(Ni-NG),其在CO2电化学还原反应中对CO 的FE 最高约为95%,此时电流密度约为11mA/cm2,并且连续反应20h后CO的FE依然可以保持在90%左右,电流密度也没有明显降低;DFT计算结果表明,较弱的CO结合能和较高的HER反应能垒是Ni-NG 催化剂上高CO 选择性的主要原因。Zhao 等[71]报道了一种新颖和通用的晶种方法,用以合成负载在不同二维材料(如石墨烯、氮化硼和二硫化钼等)上的单原子催化剂。其中氧化石墨烯上的Ni单原子(SANi-GO)在CO2电化学还原反应中表现出了极为优异的催化性能,电位为-0.63V时CO的FE高达96.5%,连续反应50h后FE仅下降到91%。
当前大部分单原子催化材料在CO2电化学还原反应中主要生成CO,而设计能将CO2还原为烃类或醇类的单原子催化剂仍然面临很大的挑战。Wang等[90]开发的负载在CeO2上的Cu单原子催化材料能以58%的FE 将CO2转化为CH4,而Jiao 等[91]将Cu 单原子固定在C3N4载体上用于电化学还原CO2,发现Cu-C3N4催化剂对C2产物表现出了较好的选择性。尽管目前报道的能有效提高C2产物选择性的单原子催化剂很少,但这些早期的探索表明,开发新型金属单原子材料、优化金属原子与载体和配位元素的相互作用对于改善产物分布或许是一种行之有效的方法。
4 结语
总而言之,得益于对反应机理的初步了解,近年来用于电化学还原CO2的催化材料不断发展,相比于传统多晶金属电极,新型金属基材料的电催化性能有了长足的进步。但较高的过电位需求、较低的产物选择性(尤其是直接还原CO2为烃类或醇类)以及不理想的反应稳定性使得金属基电催化材料离工业化应用的标准尚有差距,因而开发更高性能的电催化材料仍然是CO2电化学还原领域未来研究的主要方向之一。纳米催化剂在反应中表现优异,优化金属基纳米材料的形貌、结构、界面、化学状态和组成以及通过不同金属的合金化,调整催化剂表面与重要中间产物的结合能,是提高催化性能的有效途径。合理利用金属基复合材料中金属与载体的协同效应,在提高C2+产物选择性和反应稳定性方面很有潜力。单原子金属催化剂最近在CO2电化学反应中显示出优异的催化性能,但其研究目前还处于起步阶段,依然面临很多问题,例如为防止团聚,通常制备的单原子催化材料金属原子负载量较小,尽管转化频率值很高,但整体活性难以与纳米颗粒相比,对多碳烃类和醇类的选择性很差。因此开发新型单原子催化材料,高活性、高选择性地将CO2转化为多碳产物是一个很有前景的研究方向。此外,建立更精确的理论计算模型,结合先进的原位光谱分析,进一步深入探究电化学还原CO2的反应途径和金属基材料的催化机理,对于高效电催化材料的设计也至关重要。
