D-氨基酸氧化酶的分子改造及应用研究进展
2021-03-30居述云吴坚平杨立荣
居述云,吴坚平,杨立荣
(1 浙江大学化学工程与生物工程学院,浙江杭州310027;2 浙江大学杭州国际科创中心,浙江杭州311200)
D- 氨基酸氧化酶(D-amino acid oxidase,DAAO,EC 1.4.3.3)是一类含有黄素腺嘌呤二核苷酸(FAD)的黄素蛋白。有氧条件下,该类酶能够对映体选择性催化D-氨基酸氧化脱氢,生成亚胺酸。后者自发水解生成相应的α-酮酸和氨,见图1。反应过程中,还原型辅酶FADH2经氧气再氧化,产生过氧化氢。
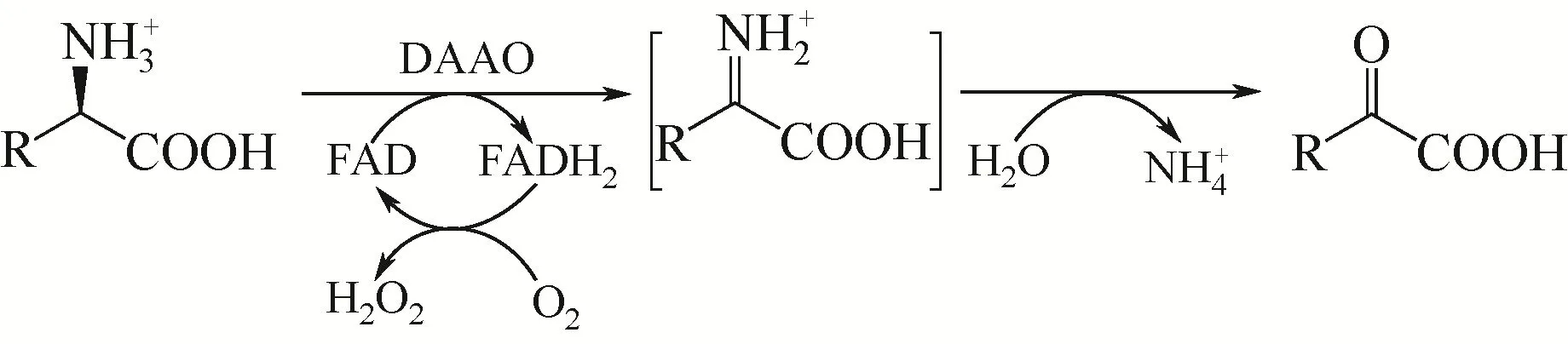
图1 DAAO催化D-氨基酸氧化脱氢反应示意图
迄今为止,已报道的DAAO主要来源于真核生物,如丝状真菌、酵母菌、哺乳动物等[1]。其中,典型的DAAO 包括猪肾(porcine kidney)来源的pkDAAO[2]、红酵母(Rhodotorula gracilis)来源的RgDAAO[3]和三角酵母(Trigonopsis variabilis)来源的TvDAAO[4]。此外,在放线菌属的原核生物中也陆续发现了DAAO[5],例如源自原玻璃蝇节杆菌(Arthrobacter protophormiae)的ApDAAO。这些不同来源的DAAO 不仅在D-氨基酸代谢过程中发挥重要作用[6],同时也为该类酶在生物技术中的应用奠定了基础。
DAAO通常具有宽泛的底物谱和高度的对映体选择性,已被成功用于多种生化反应过程,相关研究引起了国内外学者广泛的关注[7-8]。多年来,DAAO在生物催化方面的应用主要见于7-氨基头孢烷酸、手性氨基酸以及α-酮酸的合成。最近,研究人员基于该类酶的蛋白结构以及催化机制,通过蛋白质工程改造酶的催化性能,使其能够选择性催化氧化胺类化合物并用于制备手性胺,不仅成功拓展了底物特异性,而且为后续以DAAO为蛋白骨架创制新的酶类提供了重要的理论依据。
本文介绍了DAAO的蛋白结构特征及其催化机制,阐述了DAAO底物特异性和热稳定性分子改造的代表性成果。最后,总结了DAAO在生物催化与转化中的应用进展。
1 D-氨基酸氧化酶的结构特征及催化机制
1.1 D-氨基酸氧化酶的蛋白结构特征
到目前为止,蛋白质结构数据库(protein data bank,PDB)中已披露的DAAO 登录号共有39 个,包括4 个RgDAAO[9-10]、16 个pkDAAO[11-12]以及19 个人体来源的hDAAO[13]。这些结构数据都是基于X 射线晶体衍射技术而获得,分辨率在1.2~3.2Å(1Å=10-10m)。
RgDAAO、pkDAAO和hDAAO蛋白分子量大小相近,单个亚基所含有的氨基酸数目分别为368、347 和347,见表1。序列比对结果表明,pkDAAO(或hDAAO) 与RgDAAO 的序列一致性分别为28.73%(或27.73%)。
虽然RgDAAO、pkDAAO 和hDAAO 的氨基酸序列具有一定的差异,但在三维结构上却颇为相似[14-15]。一般而言,它们均为同源二聚体,每一个亚基都含有一个非共价结合的辅酶FAD。催化活性中心处与配体相互作用的氨基酸残基具有高度的保守性。例如,在RgDAAO 与D-三氟丙氨酸的复合体晶体结构(PDB登录号:1C0L[9])中,D-三氟丙氨酸的α-羧基与R285位的精氨酸残基以及Y223位的酪氨酸残基相互作用,形成了盐桥和氢键,见图2(a)。pkDAAO与苯甲酸(PDB登录号:1VE9[12])[见图2(b)]以及hDAAO与2-氨基苯甲酸(PDB登录号:2E4A[13])[见图2(c)]的复合体晶体结构解析结果表明,与配体羧基作用的正是R283 位的精氨酸残基以及Y228 位的酪氨酸残基。此外,配体的取代基团都朝向由疏水性氨基酸残基(如苯丙氨酸、甲硫氨酸、异亮氨酸、亮氨酸等)形成的口袋。DAAO蛋白晶体结构的解析不仅有利于该类酶催化机制的研究,而且为后续蛋白质工程改造酶的催化性能提供了重要的理论支撑。
1.2 D-氨基酸氧化酶的催化机制
Walsh 等[16]和Hersh 等[17]早 期 针 对DAAO 催 化的氧化脱氢反应,分别提出了“负碳离子”和“Cα-H氢转移”机制。随着pkDAAO和RgDAAO的蛋白结构相继被解析,目前普遍认可的是“氢转移”机制[9,18]。DAAO 利用辅酶FAD 在底物和O2之间进行电子和氢的转运(见图3),所催化的氧化脱氢反应由两个半反应组成。①还原半反应,底物进入DAAO 的活性中心后,以兼性离子的状态与活性中心底物结合口袋处的氨基酸残基相互作用,形成氢键、离子键等。这种状态会发生动态平衡,底物Cα上的氢转移到辅酶FAD 异咯嗪环的N(5)上,而氨基上的一个氢则释放到反应体系中,从而形成亚胺酸。该化合物不稳定,自发水解生成相应的α-酮酸和氨。②氧化半反应,还原型辅酶被O2氧化,生成过氧化氢。整个反应过程中,活性中心处的氨基酸残基并没有直接参与催化反应,仅仅与底物的结合及其保持正确定向有关[7,9,19]。
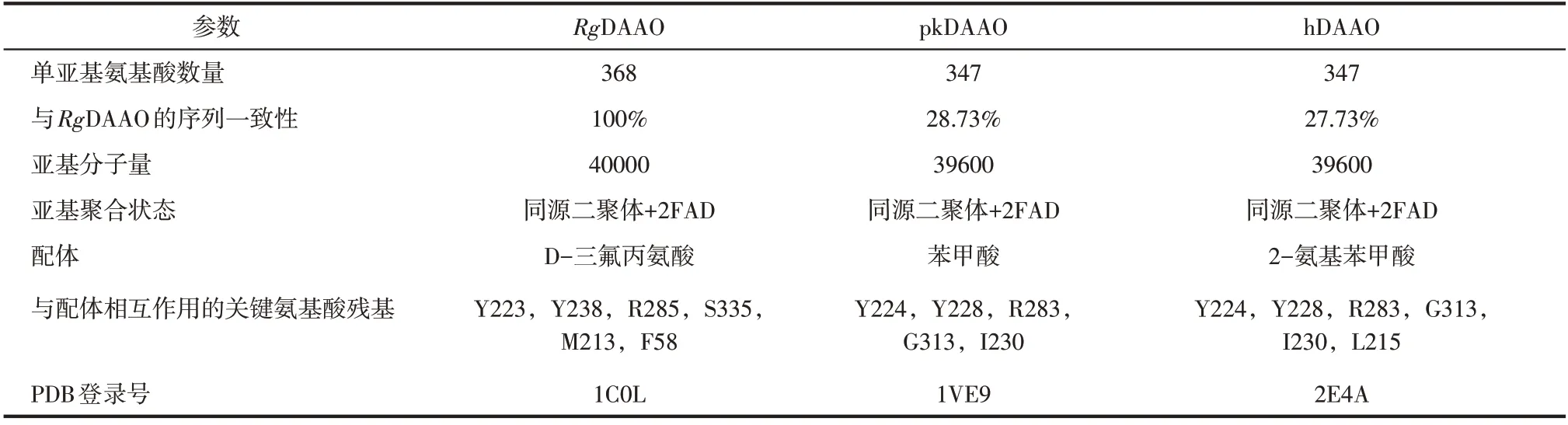
表1 RgDAAO、pkDAAO和hDAAO的基本性质比较
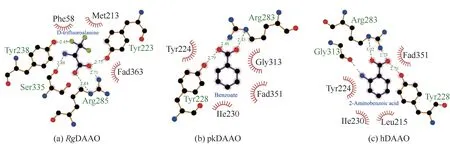
图2 RgDAAO、pkDAAO和hDAAO分别与不同配体相互作用的二维平面图
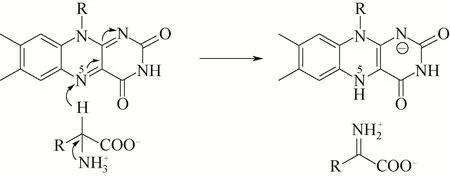
图3 DAAO催化D-氨基酸氧化脱氢的反应机制示意图
2 D-氨基酸氧化酶的蛋白质工程
2.1 D-氨基酸氧化酶底物特异性的分子改造
2.1.1 基于理性设计改变底物结合口袋处关键氨基酸残基的带电性质
RgDAAO 的底物特异性偏好中性或碱性D-氨基酸。为提高该酶对酸性氨基酸的催化活力,Pollegioni 等[20]将D-天冬氨酸(1,见表2)分别与牛肾来源D-天冬氨酸氧化酶DASPO(同源模建)和RgDAAO(PDB 登录号:1C0L)进行分子对接,选取底物结合口袋处的M213进行定点突变。结果表明M213 位甲硫氨酸残基对于RgDAAO 正确识别D-天冬氨酸侧链基团中带负电荷的羧基具有重要影响,而在DASPO 模拟结构中,与之相对应的是侧链基团带正电荷的精氨酸残基。当甲硫氨酸被精氨酸取代后,突变体M213R对D-天冬氨酸(1)和D-谷氨酸(2)的催化效率(kcat/Km值)分别是原始酶的74倍和37倍,而对D-丙氨酸(3)的催化效率却显著降低。
近年来,Asano 等[21]通过分析苯甲酸与pkDAAO的复合物晶体结构,选取与苯甲酸的羧基形成盐桥的R283作为定点饱和突变位点。当R283位侧链基团带正电荷的精氨酸残基突变为不带电荷的甘氨酸残基时,突变体R283G 转变成了具有严格(R)-构型选择性的胺氧化酶,能够对映体选择性识别(R)-α-苯乙胺(9),同时丧失了对天然底物D-苯丙氨酸的催化活力,从而将pkDAAO的底物特异性由氨基酸拓展到了胺类化合物。
上述研究表明,DAAO底物结合口袋处关键氨基酸残基的带电性质能够显著影响该酶对底物的识别能力。因此,对这些位点进行改造可以进一步拓展DAAO的底物特异性。
2.1.2 基于理性设计改变底物结合口袋处关键氨基酸残基侧链基团的大小
Pollegioni等[22]基于已有的RgDAAO晶体结构和虚拟筛选,发现M213位氨基酸残基侧链基团的大小显著影响该酶对大位阻底物的催化活力。当M213 位侧链基团较大的甲硫氨酸突变为较小的甘氨酸时,疏水性底物结合口袋的空间变大,突变体M213G对于D-1-萘基丙氨酸(4)和D-1-萘基甘氨酸(6)的催化效率相较于原始酶分别提高了8.4 倍和10.5倍。此外,对于结构刚性较大的苯甘氨酸类衍生物(5)和(7),该突变体的催化效率也有不同程度的提高[23]。
Asano 等[21]在pkDAAO 突变体R283G 的基础之上,对与苯甲酸羧基相互作用的另一位点Y228 进行突变。其中,突变体Y228L/R283G 对(R)-9 的催化活力提高到了21.5U/mg。蛋白晶体结构解析结果表明在辅酶FAD 二甲苯环的旁边产生了一个疏水口袋,从而有足够的空间能够容纳(R)-9 的苯环。尽管底物特异性发生了改变,突变体Y228L/R283G依然通过α-质子氢转移机制来进行氧化脱氢反应[24]。底物(R)-9 的Cα原子上的氢(α-质子)更靠近辅酶FAD 的N(5)原子,而(S)-9 的α-质子则朝向Y224,从而使得(R)-构型底物的氧化脱氢反应更容易进行。第一性原理计算结果表明,突变体Y228L/R283G 与(R)-9 的相互作用能比(S)-9 的相互作用能强达13kcal/mol(1kcal/mol=4.1840kJ/mol)。这些差异导致突变体Y228L/R283G 能够特异性识别外消旋体中的(R)-构型胺类化合物[25]。该课题组同时也发现pkDAAO 突变体Y228L/R283G 活性中心处F242 位氨基酸残基侧链基团的大小显著影响该酶对较大位阻底物的催化活力,见表2。当该位点的苯丙氨酸突变为侧链基团较小的异亮氨酸时,活性中心处形成了更大的疏水口袋,突变体Y228L/R283G/F242I 对(R)-1-(2-萘基)-乙胺(10)的催化活力达4U/mg,催化效率相较于突变体Y228L/R283G 提高了0.6 倍[25]。虽然突变体Y228L/R283G 能够催化氧化(R)-9 和(R)-10,但对于具有更大取代基的底物4-氯-二苯甲胺(11)却没有催化活力。Asano 等[26]根据已有的突变体Y228L/R283G晶体结构以及构效关系分析,推测活性中心处L51、I215和I230位氨基酸残基可能在该酶催化大位阻底物氧化脱氢的反应过程中起重要作用。因此,分别以突变体R283G 和Y228L/R283G 作为亲本,对上述三个位点进行定点饱和突变。突变体I230A/R283G 对 rac-11 的催化效率最高(122.7mmol/L·min),并且具有严格(S)-对映体选择性,见表2。

表2 DAAO底物特异性改造的代表性成果

续表2
由此可见,DAAO底物结合口袋的大小受关键氨基酸残基侧链基团空间位阻的影响。利用具有较小侧链基团的氨基酸代替大位阻侧链基团的氨基酸残基,可以有效扩大底物结合口袋,增强酶与底物的相互作用,进而提高该类酶的催化活力。
2.1.3 基于理性设计改变底物结合口袋处氨基酸残基的极性
到目前为止,TvDAAO的蛋白质晶体结构还未有报道。因此,通过同源建模获得较为可靠的三维模型,考察酶与底物的相互作用,在此基础上进行突变,可以获得具有目标性能的突变体。
为进一步提高TvDAAO 对头孢菌素C(8)的催化活力,Wong 等[27]基于底物8 与TvDAAO(同源模建)的分子对接结果,选取底物结合口袋处邻近底物氨基的F54 位苯丙氨酸残基作为定点突变位点。当疏水性苯丙氨酸残基变为极性相对较大的酪氨酸残基时,侧链基团中的酚羟基与底物8的氨基形成了氢键,稳定了底物在结合口袋处的构象,突变体F54Y 对底物8 的催化效率相较于原始酶提高了1倍。
2.1.4 基于定向进化改变非底物结合口袋处的氨基酸残基
理性设计主要基于酶与底物相互作用的氨基酸残基而展开改造工作。此外,通过定向进化,可以发掘一些非活性中心处的氨基酸残基对DAAO底物特异性的影响。
除上述疏水性底物结合口袋处的氨基酸残基以外,Sacchi 等[28]通过易错PCR 构建了随机突变体库,结合过氧化物酶/邻联茴香胺介导的显色法,进行高通量筛选。结果表明:位于RgDAAO 蛋白表面的L118,T60 以及Q144 位氨基酸残基能够显著影响该酶的催化活力,见表2。例如,突变体L118H对底物3和1的催化效率分别是原始酶的1.9倍和2 倍。突变体T60A/Q144R/K152E 对1 的催化效率是原始酶的5.6倍。
2.2 D-氨基酸氧化酶热稳定性的分子改造
酶的热稳定性是影响生物催化反应的重要因素。Bakke 等[29]通过易错PCR 建立突变体库,利用过氧化物酶/4-氨基安替比林介导的显色反应进行高通量筛选,获得了热稳定性显著提高的pkDAAO。55℃条件下孵育1h 后,突变体F42C 对(R)-3的残余酶活达70%以上,而原始酶仅为10%。此外,2.1.3 节中通过理性设计获得的突变体TvDAAO F54Y 不仅提高了该酶对底物8 的催化活力,而且也增强了热稳定性。55℃条件下保温30min,突变体F54Y 对8 的残余活力几乎没有影响,而原始酶却丧失了40%。
3 D-氨基酸氧化酶在生物催化中的应用
3.1 D-氨基酸氧化酶介导的两步酶法催化合成7-氨基头孢烷酸
7-氨基头孢烷酸(14)是制备半合成头孢菌素类抗生素的前体化合物。自20 世纪90 年代以来,以DAAO 和戊二酰基-7-氨基头孢烷酸酰化酶为生物催化剂,已成功建立了两步酶法催化转化头孢菌素C(8)合成14 的生产工艺(见图4),并进行了工业化生产[30]。在此过程中,DAAO催化8的D-α-氨基己二酰基团氧化脱氢生成α-酮己二酰-7-氨基头孢烷酸(12),后者经脱羧反应转化成戊二酰-7-氨基头孢烷酸(13)。最终,酰化酶催化13进行脱酰基反应生成产物14[31-32]。
3.2 D-氨基酸氧化酶催化合成手性氨基酸和胺类化合物
手性氨基酸和胺类化合物是合成众多医药、农药等高值化学品的前体化合物。例如,以(S)-1,2,3,4-四氢异喹啉-1-甲酸(16)为底物,经多步化学反应,可合成一种靶向Keap1-Nrf2 相互作用的小分子抑制剂,从而有望用于癌症、糖尿病等疾病的治疗和预防[33]。(S)-1,2,3,4-四氢异喹啉-3-甲酸(17)是降压药喹那普利的重要组成部分[34],L-2-氨基丁酸(21)是抗癫痫药物左乙拉西坦的前体化合物[35]。利用D-氨基酸氧化酶介导的去消旋化反应制备该类化合物具有外消旋底物来源简单、底物谱广泛、反应对映体选择性高、反应条件温和等显著特点。到目前为止,该方法主要有两种反应方式:①化学-酶法去消旋化;②多酶催化氧化还原级联去消旋化。

图4 两步酶法合成7-氨基头孢烷酸
3.2.1 化学-酶法去消旋化
DAAO 介导的化学-酶法去消旋化包含两步反应。①选择性生物催化反应:以外消旋氨基酸或胺为底物,利用氧化酶选择性催化其中的一个构型进行Cα-N 键氧化脱氢反应生成亚胺,另一构型保持不变。②非选择性化学还原反应:亚胺被非选择性化学还原剂转化为外消旋底物。两步反应在“一锅”反应体系中高效协同进行,见图5。亚胺(特别是链状亚胺)在水溶液中通常不稳定,易水解开环生成醛(酮)和胺。因此,反应需加入过量化学还原剂,如硼氢化钠(NaBH4)[36]、氰基硼氢化钠(NaCNBH3)[37]、氨硼烷(NH3·BH3)[38]等,以确保亚胺能够直接转化为外消旋底物,尽可能避免副产物(如酮或醇)的生成。
Chen 等[39]利用硅藻土固定化的TvDAAO 和NH3·BH3(10个当量)进行去消旋化反应,制备糖尿病药物合成中间体(S)-α-氨基-6-(2-甲基苯)3-吡啶丙酸(15),产率为68%,ee值>99%。本文作者所在课题组[40]以来源于茄病镰刀菌(Fusarium solani)M-0718 的FsDAAO 作为生物催化剂,构建了“一锅法”FsDAAO-NH3·BH3(4 个当量)去消旋化反应体系,将外消旋四氢异喹啉羧酸类化合物转化成了(S)-对映体,转化率达98%以上,产率达82%,ee 值>99%。Asano 等[21]利用pkDAAO Y228L/R283G 和NaBH4(20 个当量)进行去消旋化反应,(R)-9完全转化为(S)-对映体,产率为65%,ee值达99%。底物为rac-11 时,以pkDAAO I230A/R283G作为生物催化剂进行去消旋化反应,产物(R)-11的产率为46%,ee值为96%[26]。
该方法通常只需要一种氧化还原酶参与反应,并且所用化学还原剂来源简单。不足之处在于反应过程中通常需要加入过量的化学还原剂,不利于环境友好。反应结束后,需要额外的分离纯化步骤,用于除去剩余的化学还原剂,导致最终的产率有所下降。
3.2.2 多酶催化氧化还原级联去消旋化
多酶催化氧化还原级联去消旋化反应是在上述D-氨基酸氧化酶介导的选择性生物催化反应的基础上,利用生物催化不对称还原反应来代替非选择性化学还原反应,从而将外消旋底物(通常为氨基酸及其衍生物)转化成单一构型的产物。当底物为链状氨基酸时,其氧化脱氢产物亚胺酸通常不稳定,易自发水解生成酮酸。后者可在氨基酸脱氢酶或者转氨酶的催化下,进行不对称还原胺化反应,见图6。
Findrik 等[41]利 用 ApDAAO 和 红 球 菌 属(Rhodococcus sp.) 来源的L-苯丙氨酸脱氢酶(L-PheDH) 进行生物催化级联反应,将D-甲硫氨酸(18)几乎全部转化成了L-甲硫氨酸。Chen等[39]以TvDAAO 和脲芽孢八叠球菌(Sporosarcina ureae)来源的(S)-选择性氨基酸脱氢酶(AADH)作为催化剂进行去消旋化反应,产物(S)-15的最终分离产率为54%,ee 值>99%。此外,该作者也利用伯克霍尔德菌(Burkholderia sp.)来源的(S)-选择性转氨酶(TA)替代上述氨基酸脱氢酶,进行“一锅法”去消旋化反应,产物(S)-15 的产率达73%,ee值>99%。Qi 等[42]以外消旋氨基酸为底物,利用TvDAAO 与谷氨酸棒状杆菌(Corynebacterium glutamicum)来源的亮氨酸酸脱氢酶(CgLeuDH)进行“一锅法”去消旋化反应,制备L-氨基酸。通过批次补料,反应体系中产物L-正缬氨酸(19)的浓度达54.09g/L,转化率>96%,ee 值>99%。Green等[43]利用RgDAAO和大肠杆菌来源转氨酶EcgabT进行去消旋化反应,将DL-草铵膦(20)转化成了L-草铵膦,转化率达85%,ee 值>99%。Seo 等[44]在两相体系中,利用表达VHb-RgDAAO 融合蛋白的全细胞与表达河流孤菌(Vibrio fluvialis)来源ω-转氨酶(VfTA)的全细胞进行“一锅法”去消旋化反应,将底物rac-21 (500mmol/L)转化成了(S)-21,转化率达97%,ee 值>99%。Liu 等[45]以基因组上携带有TvDAAO 和L-PheDH 基因的酿酒酵母作为宿主细胞,将前体化合物DL-甲硫氨酸经L-甲硫氨酸转化成了S-腺苷-L-甲硫氨酸,产物积累量达10.3g/L。
不同于上述链状亚胺酸,环状亚胺酸在水溶液中较为稳定,可被(S)-选择性还原酶直接转化为(S)-构型环状氨基酸,见图7。例如,Yasuda 等[46]以外消旋哌啶酸(22)为底物,利用pkDAAO 与恶臭假单胞菌(Pseudomonas putida)KT2440 来源的还原酶PpDpkA 进行去消旋化反应,产物为(S)-22,转化率和ee 值均>99%。本文作者课题组[47]基于反应可行性及兼容性评估,成功耦合FsDAAO催化的对映体选择性氧化脱氢反应和PpDpkA介导的不对称还原反应,构建了“一锅法”多酶级联去消旋化反应体系,将底物rac-16转化成了(S)-对映体,产率为89%,ee值>99%。相较于化学-酶法去消旋化反应,该反应体系中的亚胺酸被直接转化成了目标手性化合物,从而使得整个去消旋化反应效率更高,并且反应结束后,分离纯化步骤简单,产物的收率更高。不足之处在于具有相反对映体选择性的氧化还原酶筛选困难,需要优化获得合适的反应条件,以确保整个去消旋化反应高效协同进行。
3.3 D-氨基酸氧化酶催化合成α-酮酸
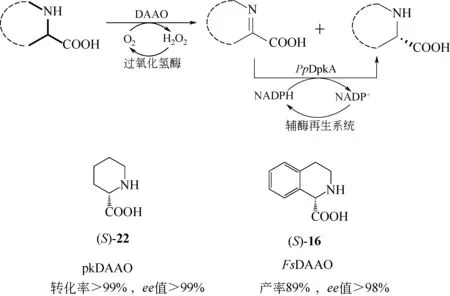
图7 DAAO介导的多酶催化级联去消旋化制备手性环状氨基酸
作为一种重要的化工原料,α-酮酸绿色高效的合成方法是近年来的热点研究内容之一。DAAO催化D-氨基酸氧化脱氢是合成相应α-酮酸的重要途径。例如,Orrego 等[48]以外消旋氨基酸为底物,利用霍乱弧菌(Vibrio cholera)来源的氨基酸消旋酶BsrV 将L-氨基酸转化成D-氨基酸,后者经TvDAAO 催化生成α-酮酸(23~25),转化率>99%,见图8。该方法的优势在于反应效率高,环境友好,并且避免了使用价格昂贵的D-氨基酸作为原料。然而,相较于L-氨基酸氧化酶或氨基酸脱氨酶催化的“一步法”合成α-酮酸[49-52],该三酶体系仍然较为复杂。
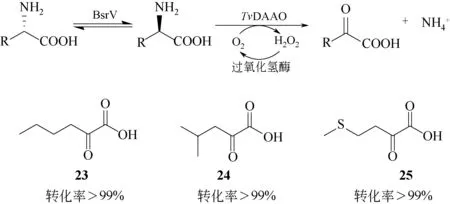
图8 DAAO催化合成α-酮酸
4 结语
DAAO作为一种重要的生物催化剂,已成功用于制备7-氨基头孢烷酸、手性氨基酸和胺类化合物等过程。活性中心处与底物相互作用的氨基酸残基主要与底物的正确定向有关。目前DAAO的分子改造工作也主要基于该处关键氨基酸残基的突变而展开,相关成果加深了该酶构效关系的理解。然而,到目前为止,能够满足生物合成需求的DAAO种类仍然相对较少,底物范围主要集中于头孢菌素C 和结构较为简单的D-氨基酸及其衍生物。后续的研究工作或许可以从以下三方面展开。
(1)挖掘具有不同底物特异性的DAAO,解析蛋白结构,结合计算化学阐明其对映体选择性及底物识别的分子机制。
(2)基于理性设计,对该类酶进行分子改造,以期获得能够催化不同非天然底物的突变体。此外,目前可用于生物催化过程的L-氨基酸氧化酶仍然稀少,如能翻转现有DAAO 的对映体选择性,使其能够催化氧化L-氨基酸及其衍生物,必将扩大该类酶的应用范围。
(3)设计新颖的化学-酶或多酶耦合催化体系,以挖掘或改造获得的DAAO作为催化剂,从而实现功能化学品生物催化合成新途径的构建。
