国家评价性抽验盐酸奥洛他定口服制剂质量分析
2021-03-18李宝霞董双涛张春泓
李宝霞,董双涛,张春泓,马 郑*
(1.山西药科职业学院,太原 030031;2.大连市药品检验所,辽宁大连 116021)
国家评价性抽验是药品主管部门为掌握全国药品的质量水平而进行的抽查检验工作〔1〕,国产药品依法定标准进行检验;然后以问题为导向,开展针对性的探索性研究。
盐酸奥洛他定(C21H23NO3·HCl)为治疗过敏性鼻炎、结膜炎的抗过敏药,也可用于治疗银屑病的瘙痒〔2-4〕,其不良反应轻微〔5〕。我国盐酸奥洛他定口服制剂分为片剂和胶囊剂,且均是仿制药。为全面系统地评价仿制药的质量优劣,我们对国产盐酸奥洛他定口服制剂进行了系统的研究,首先依照法定标准检验,然后基于风险控制的理念开展了详实的探索性研究〔6〕,全面系统地对盐酸奥洛他定口服制剂进行质量评价。
1 制剂的概况
盐酸奥洛他定为新型选择性组胺受体阻断剂。本品口服后,消除半衰期为8~12 h。该药60%~70%经肾脏以原形形式消除,17%以粪便排出。不良反应以头痛、困倦较为常见。
本次抽验涉及盐酸奥洛他定片、盐酸奥洛他定胶囊、盐酸奥洛他定滴眼液3 个剂型,收集到114 批次样品(本文只介绍口服制剂),分别来自26 个省区、直辖市。片剂和胶囊剂的内包装均为铝塑板,外包装均为纸盒,规格均为5 mg。涉及生产企业为某国外制药企业(片剂,原研制剂生产商)、某制药有限公司(片剂)、某制药有限公司(胶囊剂)。
2 法定检验结果及质量分析
2.1 法定检验结果依法定标准进行检验。本部分内容主要考察质量标准的合理性、科学性、可操作性,对数据进行对比,对法定检验中发现的问题进行剖析,并开展针对性的探索性研究。
质量标准涉及的检查项目有性状、鉴别(分别为化学鉴别、紫外鉴别、液相色谱鉴别)、含量均匀度、溶出度、有关物质及含量。所有样品的检验结果均合格,提示样品均能达到质量标准规定的各项要求。质量标准各检查项目齐全,检查方法具备可操作性,符合我国药典规定的技术要求〔7〕。存在的问题:胶囊剂的溶出度检查法采用小杯法(其他两个质量标准均采用桨法),该法是我国特有的检查法,随着我国加入人用药品注册技术要求国际协调会(ICH)并与国际制药行业发展不断融合,建议修改为桨法,与原研制剂一致。其次,胶囊剂有关物质检查法未控制特定杂质,且有关物质的限度过宽,应对质量标准进行提升。
2.2 质量分析法定检验结果虽然均合格,但从均匀度、含量、溶出度等多项统计结果来看,原研制剂数据的离散性明显小于国产制剂。以溶出度结果为例,测定结果的统计结果见图1。由图1 可知,本品溶出完全,在15 min 时,片剂已完全溶出(近100%)说明进口制剂的工艺水平优于国产制剂。从有关物质的数据来看,国产片剂的杂质水平优于或与原研制剂相当。胶囊剂的杂质水平明显偏大,提示存在一定安全性风险。
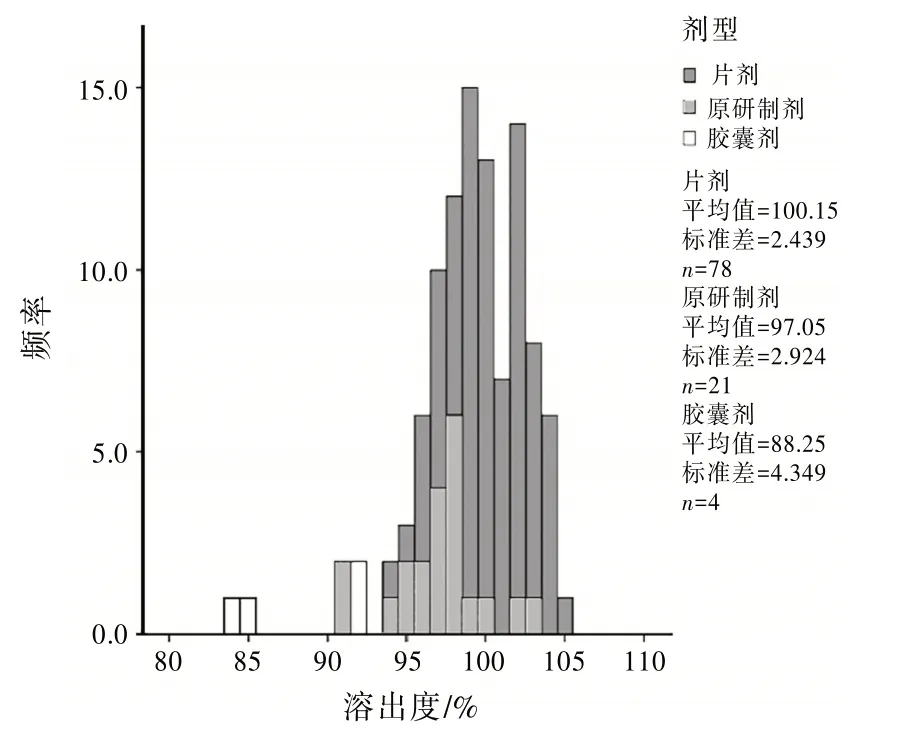
图1 溶出度检查频数分布图
3 探索性研究
本研究以风险控制的理念,结合法定检验中发现的问题,从药品的安全性、有效性以及标准的科学、合理性三个方面,对贯穿于药物全生命周期中可能存在的高风险因素展开与原研制剂的对比研究,以期最大程度保障人民群众用药安全。
3.1 安全性评价安全性评价基于与原研制剂的对比研究并考察了贯穿于药品整个生命周期的高风险因素。在风险因素的设置上,重点考虑到我国国情及监管方向,同时关注美国食品药品监督管理局(FDA)、欧洲药品管理局(EMA)近年来重点警示的内容。
3.1.1 基因毒性杂质的研究 基因毒性杂质本身能直接或间接损伤细胞DNA,产生基因突变或体内诱变,具有致癌可能或者倾向。由于毒性强烈,应进行严格控制〔8〕。近期,业内爆出“缬沙坦”事件,其原料中存在基因毒性杂质N-亚硝基二甲胺。
本品从合成工艺的理论上分析,也可能存在该基因毒性杂质。因此,开展了可能存在的基因毒性杂质的研究。据文献〔9〕报道,N-亚硝基二甲胺可采用气相色谱-质谱联用(GC-MS)法,以特征离子m∕z74,选择m∕z15、42、43作为限定离子,进行定量分析。
气相色谱:色谱柱:SH-RXI-5sil(30 m × 0.25 mm×0.25 μm),甲基(5%苯基)聚硅氧烷为固定相;程序升温,初始温度为150 ℃,保持5 min,再以40 ℃∕min 升至220 ℃,保持15 min。进样口280 ℃,流速1.0 mL∕min,电离方式EI,溶剂延迟5 min,分流进样,进样量1 μL。
质谱条件:电子轰击离子源,电离能力70 eV,离子源温度250 ℃,四级杆温度150 ℃。
测定法:分别取国产片剂,加N,N-二甲基甲酰胺(DMF)制成浓度为1 mg∕mL的溶液。
结果:原料中未发现该基因毒性杂质。
结论:本品国产片剂原料未发现N-亚硝基二甲胺。通过工艺、结构分析,本品工艺及产品中不存在其他警示性结构基团。原料未发现安全性风险。
3.1.2 有关物质研究 依据法定检验,国产制剂有关物质检查方法存在诸多不足:杂质控制不严、定量方法不先进。在探索性研究中,以问题为导向,开展针对性的探索性研究,建立了新的有关物质检查方法,同时测定制剂(片剂、胶囊剂)中4种已知杂质的含量。在拟定条件下,有关物质色谱图见图2,在此条件下分离出更多的杂质,并实现了4 种已知杂质的控制。
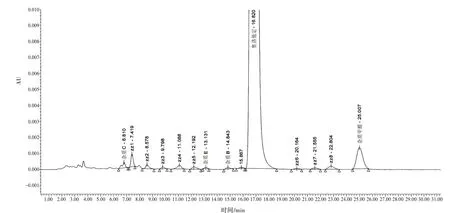
图2 拟定条件下胶囊剂有关物质色谱图
对比研究数据结果见表1。由表1可知,国产片剂质量较好,与原研制剂杂质谱一致;国产胶囊剂的杂质水平相对较高。
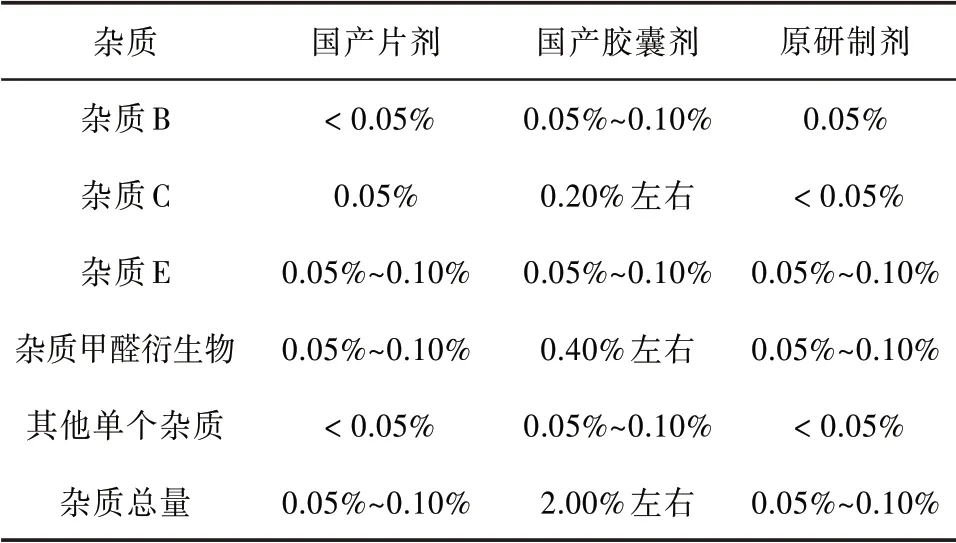
表1 对比研究结果
3.2 有效性评价有效性评价研究内容见表2。经文献调研本品为BCS1类或3类的药物。在现行质量标准中,溶出度检查法是以水为介质,并规定在15 min 内释放85%以上。通过进一步研究,本品片剂以水为介质,桨法50 转的条件下迅速崩解,基本在5 min 内可释放85%以上。胶囊剂在小杯法中的释放稍慢,但是在15 min 内也可达到80%的溶出量。依据FDA〔10〕发布的相关指导原则,本品可视为速释制剂,溶出不为本药物吸收的“限速步骤”。因此,本研究将溶出度检查项视为中低风险因素。但是为更好反应制剂厂家工艺水平,我们开展了3 种条件下溶出曲线的对比研究,发现基本一致。由于胶囊剂检查法为小杯法,我们又开展了与桨法的对比研究,发现桨法同样适合本品溶出度检查,因此建议更改为桨法,即与原研制剂溶出方法一致。对于本类药物,晶型与粒度均为低风险因素。对于晶型,经文献调研,未检索到晶型与药效之间的报道。
3.3 标准的科学、合理性
3.3.1 原料药标准 在项目的设置上,我国盐酸奥洛他定原料质量标准与国外药典标准基本一致。在限度的设立上,我国标准与国外标准主要差别在重金属和有关物质项。重金属检查项,国外药典如《美国药典》〔11〕限度均为10 ppm,严于我国20 ppm的限度。对于口服片剂品种,20 ppm 的限度符合我国现阶段对口服制剂的技术要求。基于风险控制的理念,从安全性、剂型、给药途径等综合考虑,我们认为基本合理〔12〕。

表2 有效性评价研究内容
3.3.2 制剂的质量标准 我国制剂质量标准的缺陷有:
(1)片剂的质量标准:单个杂质限度较宽;定量方法采用主成分自身对照法。其次,含量测定项下检测波长设置不合理。
(2)胶囊剂的质量标准:单个杂质限度较宽,未控制特定杂质;溶出度方法为小杯法。含量测定方法采用四氢呋喃、离子对试剂等多种试剂,繁琐复杂,耐用性差。检测波长混乱,如含量测定采用206 nm(流动相为四氢呋喃含离子对试剂),溶出度采用300 nm(介质为水),有关物质采用245 nm,而主药及杂质的最大吸收波长均在270~299 nm处。
4 结论
本研究开展了盐酸奥洛他定口服制剂国家评价性抽验。按法定标准检验,该品种合格率为100.0%。基于风险控制的理念开展探索性研究,内容包括基因毒性杂质研究,有关物质研究、晶型和粒度的调研等。经详实的研究,结果表明我国片剂总体质量较好,胶囊剂质量需进一步提高,并建议厂家提升原料和制剂的有关物质控制方法,修改胶囊剂的溶出度检查法。
