TJP2 基因突变致进行性家族性肝内胆汁淤积症3 例报告及文献复习
2021-02-26王怡仲
葛 婷 王怡仲 张 婷
上海市儿童医院 上海交通大学附属儿童医院消化科(上海 200062)
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis,PFⅠC)最早于1969年报道,是一组罕见的异质性常染色体隐性遗传疾病,发病率为1/100000至1/50000[1]。PFⅠC通常在婴儿期或儿童期起病,临床上以严重肝内胆汁淤积为主要特征,多表现为渐进性黄疸、持续性胆汁淤积、瘙痒和生长发育障碍。PFⅠC可引起早发的进行性肝损伤,若未及时治疗,持续的胆汁淤积可迅速导致终末期肝病甚至患儿死亡[1-2]。根据致病基因不同,PFⅠC 可分为1~6型。PFⅠC 1型也称Byler病,是由编码胆小管转运蛋白的ATP8B1基因(定位于染色体18q21)变异所致;PFⅠC 2 型由胆盐外运泵基因ABCB 11(位于染色体2q24)变异引起;PFⅠC 3型由编码多重耐药蛋白3型的ABCB4基因(位于染色体7q21.1)变异引起;PFⅠC 4型由紧密连接蛋白2(tight junction protein 2,TJP2)基因变异引起;2016 年首次报道的PFⅠC 5 型由编码FXR的NR1H4基因(位于常染色体12q23.1)变异所致;PFⅠC 6型由编码肌球蛋白5B的MYO5B基因(位于常染色体18q21.1区域)变异所致。现报告2017至2019年上海市儿童医院诊治的3 例由TJP 2基因变异导致的PFⅠC 4型患儿的临床资料,并复习相关文献,以提高对PFⅠC 4和TJP2变异相关疾病的认识。
1 临床资料
例1,女,1 岁11 月龄,因皮肤黄染1 年余就诊。患儿为G 1 P 1,30 周早产,出生体质量1 800 g,父母非近亲婚配,母孕期和患儿出生时无不良事件,否认家族遗传疾病史。入院体格检查:体温36.9 ℃,心率119 次/min,呼吸22 次/min,血压92/67 mmHg,体质量9.8 kg(P10~P25),身长76 cm(<P3);颜面、躯干和四肢皮肤轻度黄染,巩膜中度黄染,手、足心无黄染,双耳尖,眼窝深,心肺无异常;腹软,肝肋下4.5 cm,质软,脾肋下2 cm,质软;四肢肌力、肌张力无异常,神经系统检查无异常。入院后实验室检查:血清总胆红素(TB)70.08 μmol/L,直接胆红素(DB)35.00 μmol/L,丙氨酸氨基转移酶(ALT)147 U/L,天冬氨酸氨基转移酶(AST)112 U/L,总胆汁酸(TBA)203 μmol/L;γ-谷氨酰转肽酶(GGT)32 U/L。磁共振胰胆管造影(MRCP)无异常。腹部超声示肝肿大。胸片示胸椎无异常。心脏超声未见异常。见表1。
例2,男,4月龄,因皮肤黄染1周余就诊。患儿为G1P1,足月顺产,出生体质量3 260 g,父母非近亲婚配,母孕期和患儿出生时无不良事件,否认家族遗传疾病史。入院体格检查:体温36.6 ℃,心率132次/min,呼吸30次/min,血压82/50 mmHg,体质量7.9kg (P75~P90),身长68.5 cm(>P97);颜面、躯干和四肢皮肤轻到中度黄染,巩膜中度黄染,手、足心无黄染,无特殊面容;心肺无异常;腹软,肝肋下3 cm,质软,脾肋下未及;四肢肌力、肌张力无异常;神经系统检查无异常。实验室检查:TB 199.66 μmol/L,DB 119.00 μmol/L,ALT 68 U/L,AST 98 U/L,TBA 203 μmol/L,GGT 25 U/L。腹部及盆腔增强CT未见明显异常。胆道MRⅠ及MRCP未见明显异常。腹部超声提示肝胆系统未见明显异常。胸片胸椎正常。心脏超声未见异常。见表1。
例3,男,2 个月15 天,因皮肤黄染2 月余就诊。患儿为G 1 P 1,足月剖宫产,出生体质量3 950 g,父母非近亲婚配,母孕期和患儿出生时无不良事件,否认家族遗传疾病史。入院体格检查:体温36.5 ℃,心率128次/min,呼吸40次/min,血压83/46 mmHg,体质量5.9 kg (P25~P50),身高46 cm(<P3);颜面、躯干和四肢皮肤轻度黄染,巩膜轻度黄染,手、足心无黄染,无特殊面容;心肺腹无异常;四肢肌力、肌张力无异常;神经系统检查无异常。入院实验室检查:TB 60 μmol/L,DB 50 μmol/L,ALT 158 U/L,AST 110 U/L,TBA 130 μmol/L,GGT 21U/L。腹部超声示肝胆系统未见明显异常。上腹部MRⅠ及MRCP未见明显异常。胸片胸椎无异常。心脏超声未见异常。

表1 3例PFIC 4患儿临床特征
因患儿表现为持续性黄疸、胆汁淤积、GGT 正常或偏低,提示低GGT 胆汁淤积症。为进一步明确病因,经医院医学伦理审核,并征得父母知情同意后,行遗传性肝病基因包(含396 个已报道和遗传性代谢性肝病相关的基因)外显子基因检测。结果显示,3 例患儿均发现TJP 2基因复合杂合变异(表1),例1 变异位点为c.2448+1 G>C 和c.2639 delC(p.T880Sfs*12)[3],例2为c.1973delG (p.R658Kfs*8)和c.3001C>T (p.R1001*),见图1,例3为c.2909G>A(p.Arg970Gln)和c.3407+3A>G(p.?),见图2。3例患儿的复合杂合变异均分别遗传自父母(均为杂合状态)。例1 的c.2448+1 G>C 位于16 号外显子的剪切位点,c.2639delC(p.T880Sfs*12)为移码变异,导致mRNA 转录在17 号外显子终止;例2 的c.1973 delG(p.R658Kfs*8)为移码变异,c.3001C>T(p.R1001*)为无义变异;例3 的c.2909 G>A(p.Arg 970 Gln)和c.3407+3 A>G(p.?)可能影响mRNA 剪接。未发现ATP 8 B 1、ABCB 11、ABCB 4、NR 1 H 4等与PFⅠC相关的基因变异。
结合临床及基因检测结果,3例患儿诊断为PFⅠC 4型。予口服熊去氧胆酸25 mg/(kg·d)、复方甘草酸片2.5 mg/(kg·d)、维生素K、维生素A、维生素D、维生素E治疗。住院期间肝酶指标逐渐下降,黄疸好转,但胆汁酸仍有反复波动。出院后其中1例女性患儿定期随访中,目前肝酶指标正常,黄疸消失,但胆汁酸仍有波动,生长发育指标已达同龄儿童水平。余2 例患儿未定期随访,未能追述到患儿的进一步情况。
2 讨论
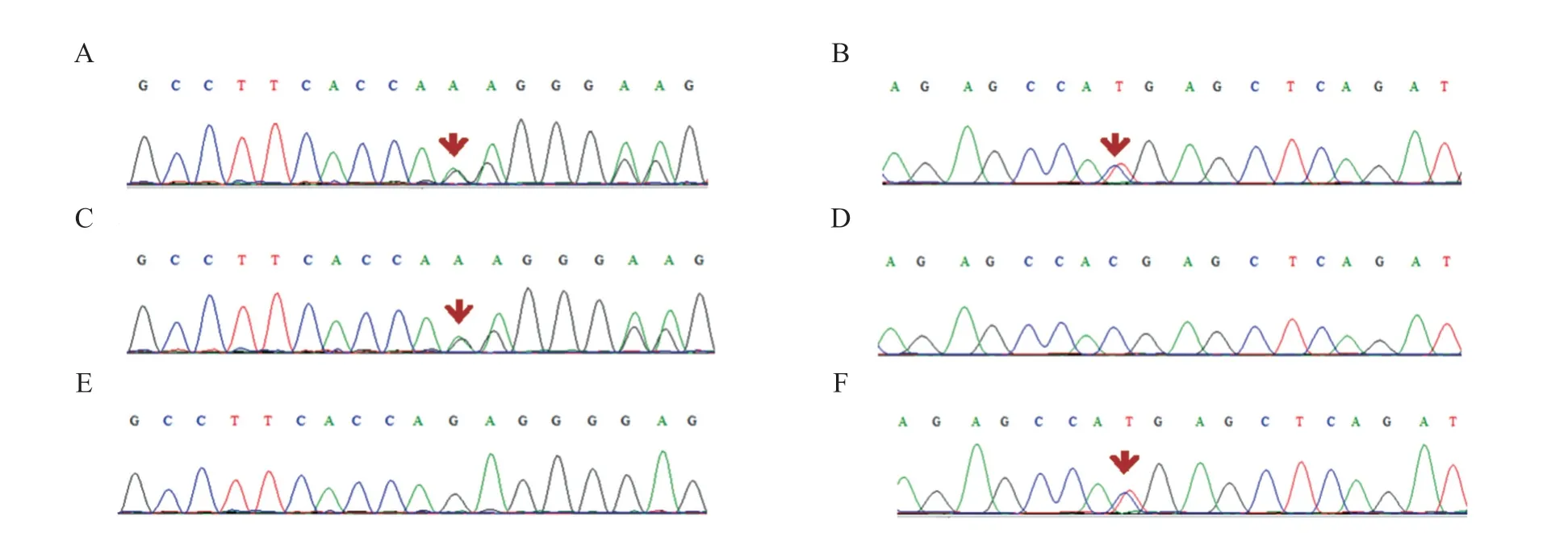
图1 例2 患儿及父母TJP2 基因Sanger 测序图

图2 例3 患儿及父母TJP2 基因Sanger 测序图
PFⅠC 4 型患儿出生后不久发病,γ-GT 正常,胆汁淤积明显,迅速发展成肝硬化,患肝细胞癌的风险增加。TJP2基因位于9号染色体的长臂上,编码TJP2蛋白,也称为zona ocdudens 2(ZO-2)[4]。TJP2蛋白是细胞间紧密连接复合体的重要组成部分,在细胞-细胞间许多位点上连接,与膜蛋白和细胞骨架蛋白相互作用,形成细胞间的紧密连接,也称闭锁小带。研究证实,TJP2变异可导致严重的胆汁淤积性肝病[2]。日本科学家在2014年首次报道12例严重胆汁淤积性肝病患儿携带多种TJP2基因纯合变异导致PFⅠC,并归类为PFⅠC 4[5]。这些基因变异导致TJP2蛋白定位失败,紧密连接结构破坏,毛细胆管中的胆汁成分渗漏进入肝实质,引起胆汁淤积。电镜下发现,存在TJP2基因变异的PFⅠC 4 患儿肝细胞间的紧密连接变长并缺少密集的闭锁小带[6]。此外,有研究报道,TJP 2变异儿童在不同年龄阶段可能会出现不同程度的肝酶、胆汁酸升高和先天性肝纤维化,甚至进展为肝细胞癌[7-8]。已有报道3 例携带TJP 2变异的儿童肝癌患者,均表现为新生儿期黄疸及正常范围的GGT,提示由PFⅠC 4进展为肝癌[9-10]。本研究小组于2019年报道1例由TJP 2复合杂合变异引起的PFⅠC 4 儿童病例[3]。本文总结包括2019 年已报道的1 例以及其后2 例新诊断的TJP2基因复合杂合变异导致的PFⅠC 4患儿的临床特征,显示3例患儿均为婴儿期起病,存在黄疸、胆汁酸升高,表现为肝内胆汁淤积症和正常范围的GGT,其中2 例患儿存在生长发育落后。基因检测发现3 例患儿存在不同位点的TJP 2基因复合杂合突变。最新研究显示,TJP 2基因变异也可引起晚发型的成人PFⅠC 4[11]。
研究证实,TJP 2变异导致的疾病具有明显异质性,与多种肝脏内和肝脏外的疾病相关。检索PubMed 关于TJP 2变异引起儿童疾病的文献,已有多个研究报告引起儿童严重肝脏疾病的TJP2纯合变异(表2)。2003 年报道在家族性高胆烷血症(familial hypercholanemia,FHC)的阿米什后裔患者中发现TJP 2变异[12]。FHC 是非典型的肝脏疾病,通常表现为血清胆汁酸显著升高,其余肝功能指标多正常,临床表现上患者多数有瘙痒、脂溶性维生素缺乏的表现。TJP2基因变异导致严重的肝脏疾病,同时还可伴有一些明显的肝脏外的病变。有研究证实,TJP 2变异与遗传性耳聋有关[13-15]。一项韩国人群研究发现,TJP 2基因变异可导致常染色体显性遗传性非综合征型耳聋(autosomal dominant non-syndromic hearing loss,ADNSHL)[15]。也有研究显示,TJP2的过表达和年龄相关的进行性非综合征性听力损失相关[13]。另外,在妊娠肝内胆汁淤积症(intrahepatic cholestasis of pregnancy,ⅠCP)成年患者中也发现了TJP2的杂合变异[16]。这些研究提示TJP 2变异临床表型的多样性。动物研究表明,缺乏ZO-2 蛋白导致小鼠早期胚胎致死,提示器官和物种之间的连接复合体的冗余度存在差异[17]。
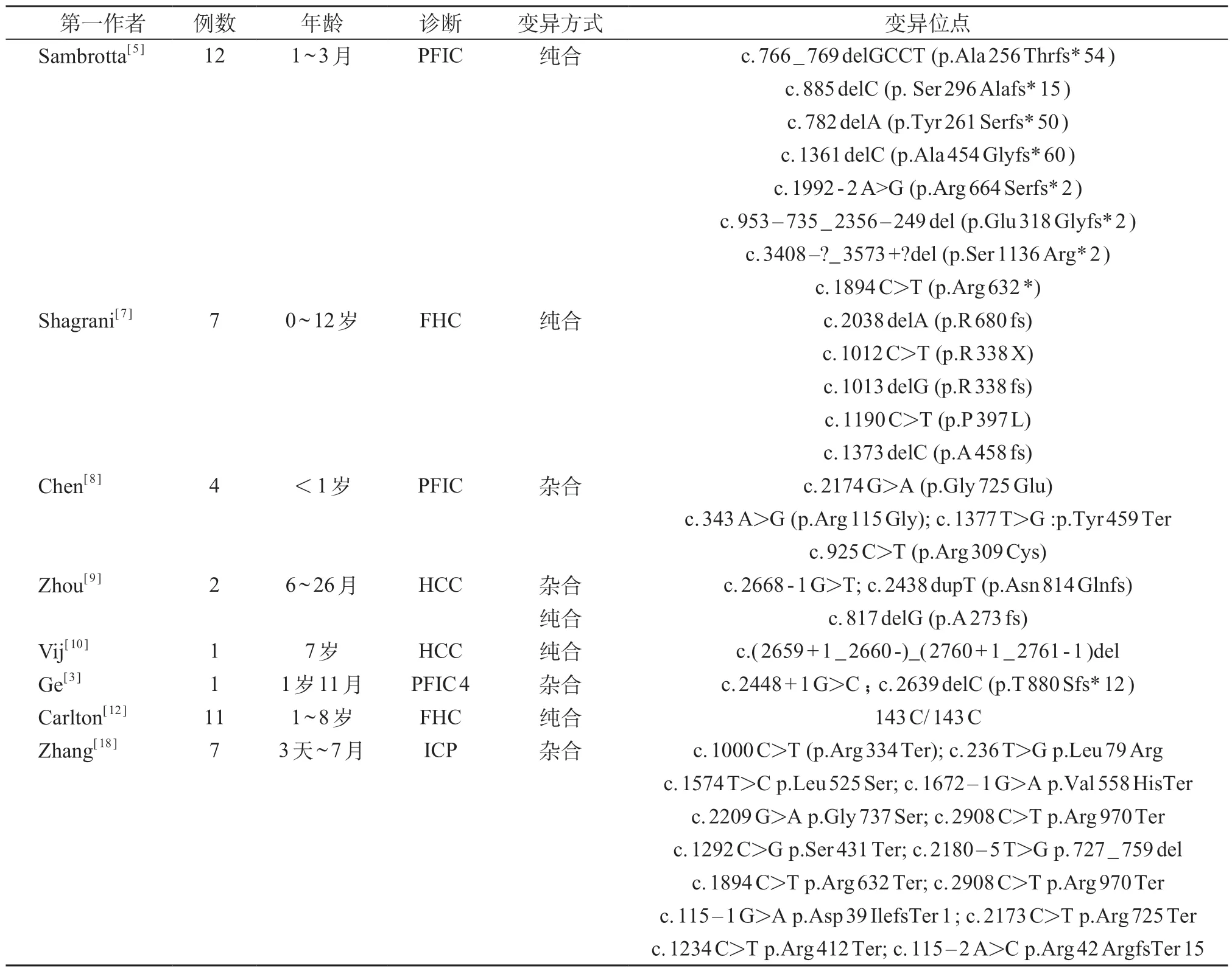
表2 儿童TJP2变异导致疾病的文献总结
综上,与其他基因变异导致的肝内胆汁淤积症一样,TJP2变异导致的肝脏疾病在儿童时期临床表现呈多样性。建议在评估有瘙痒、黄疸和正常的血清GGT儿童时考虑TJP2变异[18]。由于TJP2变异可能是儿童早期发生原发性肝癌的危险因素,因此应密切随访监测PFⅠC 4患儿情况。
