内生真菌Paraconiothyrium brasiliense代谢产物β-咔啉生物碱及其黄嘌呤氧化酶抑制活性研究
2021-02-01周继慧闫鑫磊刘朝霞刘呈雄
周继慧,闫鑫磊,田 梦,李 慧,刘朝霞,刘呈雄,2*,邹 坤*
1三峡大学生物与制药学院,天然产物研究与利用湖北省重点实验室;2三峡大学生物与制药学院,中国轻工业功能酵母重点实验室,宜昌 443002
川芎哚(川芎Ⅲ号碱,perlolyrine)是中药川芎的有效活性β-咔啉生物碱之一,具有很强的药理活性,对冠心病和脑血管病有一定的疗效,但其在体内代谢迅速、排泄较快、生物利用度低及药效维持时间较短,经化学改造后的川芎哚类似物在抗血小板聚集和体外抗血栓形成作用明显强于川芎哚和川芎嗪,且作用时间明显延长[1-4],然而植物来源的川芎哚含量较少且其类似物种类较少,且目前为止,微生物来源的川芎哚,非常罕见,只有Jishengellaendophytica[5]和真菌Fusariumequiseti[6]、Trametestrogii[7]能产川芎哚。因此,有必要通过各种有效途径获取川芎哚类似物,研究其构效关系和作用机制,从中筛选出高效、低毒、作用时间长的治疗心血管疾病的新型药物。
本课题组前期从中华剑角蝗肠道中分得一株类壳小圆孢真菌Paraconiothyriumbrasiliense(P.brasiliense),经发酵后分离,首次发现该真菌可生产天然川芎哚[8,9],但其产量很低,本课题通过前体添加介导该株真菌的次级代谢产物调节,结果使得川芎哚的产量得到明显提高,且生成了10个β-咔啉生物碱,文献报道化合物3具有较强的黄嘌呤氧化酶抑制活性[10],近年来,从动物实验到人体研究都证实了黄嘌呤氧化酶与心血管疾病密切相关,黄嘌呤氧化酶抑制剂可应用于多种心血管疾病的治疗[11]。本研究通过测定10个所分离的生物碱对黄嘌呤氧化酶的抑制活性,以别嘌呤醇作为阳性药,选择出抑制活性较好的化合物,研究其构效关系,对于川芎哚及其类似物的产量和种类提供了思路,并为开发出具有优良利用价值的黄嘌呤氧化酶抑制剂提供了参考依据。
1 仪器与材料
1.1 仪器与试剂
Bruker AV 400 MHz核磁共振波谱仪(瑞士布鲁克公司);Dionex Ultimate 3000 型高效液相色谱仪(美国戴安公司);Waters 1525EF高效液相色谱(美国Waters公司);Venusil XBP C18色谱柱(250 mm×10 mm,10 μm,半制备型;250 mm×4.6 mm,5 μm,分析型,天津博纳艾杰尔科技有限公司);低温冷冻干燥机(美国Labconco公司);正相色谱硅胶(烟台化学工业研究所);电子天平(上海民桥精密仪器有限公司);紫外分光光度计(上海谱元仪器有限公司);黄嘌呤氧化酶(signa);别嘌呤醇(Ruibio);黄嘌呤氧化酶试剂盒(南京建成生物工程研究所);高效液相用乙腈、甲醇均为色谱纯(美国Tedia公司),水为三蒸水;其他试剂均为分析纯(国药集团化学试剂有限公司)。
1.2 菌种与材料
本实验室前期从中华剑角蝗肠道中分得一株类壳小圆孢真菌Paraconiothyriumbrasiliense(P.brasiliense,Genbank Accession No.KM880019),经发酵后分离,首次发现该真菌可生产天然川芎哚(已授权中国发明专利,专利号:ZL201610408151.X)。
PDA培养基(g/L):新鲜的去皮马铃薯200.0 g,葡萄糖 20.0 g,琼脂20.0 g,自然pH,0.1 MPa,灭菌20 min。
PDB培养基(g/L):新鲜的去皮马铃薯200.0 g,葡萄糖 20.0 g,自然pH,0.1 MPa,灭菌20 min。
2 实验方法
2.1 发酵、提取和分离
将采用PDA培养基活化好的菌种接种于PDB培养基,在28 ℃,180 rpm条件下分批次加入终浓度为1.0 mM的前体L-色氨酸,继续培养20天后收集发酵液和菌丝体,采用乙酸乙酯萃取发酵液3次,菌丝体在45 ℃条件下烘干,加入丙酮浸泡过夜,超声(45 ℃、60 W)提取30 min,反复提取3次,合并所有浸提物总重量为15 g。总提取物进行正相硅胶(200~300目)柱层析分离,采用氯仿-甲醇=20∶1(体积比)横梯度洗脱,共得到50个流份。进行TLC检测,展开体系为:氯仿-甲醇=15∶1(体积比),在365 nm紫外灯下观察荧光,再用改良碘化必钾喷雾显色,观察有无橙色斑点,经TLC检测发现第25个到第30个流份,共6个流份中具有紫外365 nm荧光显色和改良碘化必钾喷雾显橙色的斑点,合并这6个流份。合并后经制备型和半制备型高效液相(HPLC)反复精制纯化。最终分离得到化合物1(200 mg)、2(3.0 mg)、3(2.5 mg)、4(3.1 mg)、5(3.4 mg)、6(3.6 mg)、7(3.3 mg)、8(3.4 mg)、9(2.0 mg)、10(2.3 mg)。化合物结构均采用质谱(MS)、核磁共振(NMR)波谱方法鉴定,并与文献对照确定化合物1~10的结构。
2.2 黄嘌呤氧化酶抑制活性

3 实验结果
3.1 化合物的结构鉴定
采用前体介导调控技术,以L-色氨酸作为前体添加后,川芎哚的含量显著提高,且通过TLC 365 nm显色和生物碱显色反应以及HPLC DAD紫外扫描,发现一系列与川芎哚紫外吸收类似的峰如图1所示,最终分离得到10个化合物如图1所示。
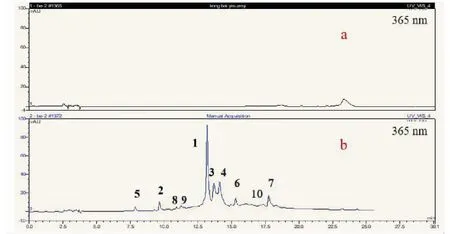
图1 添加L-色氨酸后菌株发酵液HPLC分析Fig.1 The HPLC analysis of fermentation broth of L-tryptophan added into the strain注:a:未添加L-色氨酸;b:添加L-色氨酸。Note:a:No L-tryptophan added,b:L-tryptophan added.
化合物1淡黄色针状晶体;mp.185~187 ℃;UVλmax= 237,252,275,292,364,378 nm;EI-MS:m/z264 [M]+;1H NMR(400 MHz,DMSO-d6)δ:11.30(1H,s,H-9),8.36(1H,d,J= 5.1 Hz,H-3),8.26(1H,d,J= 7.8 Hz,H-5),8.07(1H,d,J= 5.1 Hz,H-4),7.79(1H,d,J= 8.2 Hz,H-8),7.59(1H,t,J= 7.0 Hz,H-7),7.28(1H,t,J= 7.9 Hz,H-6),7.20(1H,d,J= 3.4 Hz,H-3′),6.58(1H,d,J= 3.4 Hz,H-4′),4.65(2H,s,H-6′);13C NMR(100 MHz,DMSO-d6)δ:156.7(C-2′),152.1(C-5′),140.9(C-1),138.2(C-3),133.1(C-8a),130.5(C-4b),129.4(C-6),128.4(C-9a),121.6(C-5),120.6(C-4a),119.7(C-7),113.6(C-4),112.5(C-3′),109.6(C-4′),109.0(C-8),55.9(C-6′)。该化合物由苯环、吡啶环和呋喃环构成,经查阅文献,该化合物的碳谱和氢谱数据与文献[13]报道数据一致,故鉴定该化合物为川芎哚。
化合物2淡黄色粉末;mp.190~191 ℃;UVλmax=237,252,275,292,364,378 nm;EI-MS:m/z284 [M]+;1H NMR(400 MHz,DMSO-d6)δ:11.51(1H,s,H-9),8.24(1H,d,J=5.2 Hz,H-3),8.20(1H,d,J=7.8 Hz,H-5),8.00(1H,d,J=5.2 Hz,H-4),7.70(1H,d,J=8.2 Hz,H-8),7.52(1H,ddd,J=8.2,7.2,1.0 Hz,H-7),7.22(1H,ddd,J=7.8,7.2,0.5 Hz,H-6),5.32(1H,br t,J=6.5 Hz,H-1′),3.56(1H,ddd,J=9.2,5.6,2.8 Hz,H-3′),3.51(1H,m,H-5a′),3.33(1H,m,H-4′),3.33(1H,m,H-5b′),2.30(1H,ddd,J=14.0,6.3,2.8 Hz,H-2a′),1.98(1H,ddd,J=14.0,9.2,7.5 Hz,H-2b′);13C NMR(100 MHz,DMSO-d6)δ:148.0(C-1),140.6(C-8a),136.5(C-3),132.8(C-9a),128.2(C-4a),127.8(C-7),121.4(C-5),120.5(C-4b),119.0(C-6),113.5(C-4),112.4(C-8),75.0(C-4′),72.0(C-1′),70.2(C-3′),63.1(C-5′),39.4(C-2′)。该化合物由苯环、吡啶环和1′,2′-二去氧吡喃核糖构成,经查阅文献,该化合物的碳谱和氢谱数据与文献[14]报道数据一致,故鉴定该化合物为1-(1′,2′-dideoxy-α-D-ribopyranosyl)-β-carboline。
化合物3淡黄色粉末;mp.231~233 ℃;UVλmax=217,275,364,378 nm;EI-MS:m/z308 [M]+;1H NMR(400 MHz,DMSO-d6)δ:11.68(1H,s,H-9),8.85(1H,s,H-4),8.43(1H,d,J=8.0 Hz,H-5),7.83(1H,d,J=8.2 Hz,H-8),7.67(1H,t,J= 7.8 Hz,H-7),7.44(1H,d,J=3.3 Hz,H-4′),7.37(1H,t,J=7.5 Hz,H-6),6.64(1H,d,J=3.3 Hz,H-3′),4.67(2H,s,H-6′);13C NMR(100 MHz,DMSO-d6)δ:167.3(C-7′),158.6(C-2′),151.5(C-5′),141.9(C-8a),132.9(C-1),132.3(C-9a),130.3(C-3),129.4(C-7),122.4(C-5),121.4(C-4b),121.0(C-4a),119.3(C-6),116.1(C-4),113.3(C-8),111.5(C-4′),109.7(C-3′),56.4(C-6′)。该化合物的碳谱和氢谱数据与文献[15]报道数据一致,故鉴定该化合物为flazin。
化合物4淡黄色粉末;mp.225~227 ℃;UVλmax=238,254,272,295,366,375 nm;EI-MS:m/z302 [M]+;1H NMR(400 MHz,DMSO-d6)δ:11.45(1H,s,H-9),8.24(1H,d,J= 5.2 Hz,H-3),8.21(1H,d,J=7.9 Hz,H-5),8.00(1H,d,J= 5.2 Hz,H-4),7.71(1H,d,J=8.7 Hz,H-8),7.52(1H,t,J=7.4 Hz,H-7),7.22(1H,t,J=7.4 Hz,H-6),5.30(1H,m,H-1′),3.51(1H,m,H-3′),3.48(1H,overlapped,H-5′a),3.34(1H,overlapped,H-5′b),3.33(1H,m,H-4′),3.31(1H,m,H-2′a),1.97(1H,m,H-2′b);13C NMR(100 MHz,DMSO-d6)δ:148.4(C-1),141.0(C-8a),137.0(C-3),133.1(C-9a),128.7(C-4a),128.2(C-7),121.8(C-5),120.9(C-4b),119.4(C-6),114.0(C-4),113.0(C-8),75.5(C-4′),72.6(C-1′),70.6(C-3′),63.9(C-5′),40.9(C-2′)。该化合物的碳谱和氢谱数据与文献[16,17]报道数据一致,故鉴定该化合物为tangutorid E。
化合物5淡黄色粉末;mp.202~204 ℃;UVλmax= 260,282,305,380 nm;EI-MS:m/z210 [M]+;1H NMR(400 MHz,DMSO-d6)δ:11.90(1H,s,H-9),8.50(1H,d,J= 4.7 Hz,H-3),8.42(1H,d,J= 5.0 Hz,H-4),8.30(1H,d,J= 7.8 Hz,H-5),7.80(1H,d,J= 8.0 Hz,H-8),7.60(1H,ddd,J= 8.2,7.2,1.0 Hz,H-7),7.30(1H,ddd,J= 8.2,7.2,1.0 Hz,H-6),2.80(3H,s,H-2′);13C NMR(100 MHz,DMSO-d6)δ:201.9(C-1′),142.3(C-1),137.8(C-3),136.2(C-8a),134.3(C-9a),131.3(C-4b),129.3(C-6),122.2(C-5),120.7(C-4a),120.3(C-7),120.0(C-4),113.5(C-8),26.4(C-2′)。该化合物的碳谱和氢谱数据与文献[18,19]报道数据一致,故鉴定该化合物为1-acetyl-β-carboline。
化合物6淡黄色粉末;mp.234~236 ℃;UVλmax= 220,282,308,380 nm;EI-MS:m/z268 [M]+;1H NMR(400 MHz,DMSO-d6)δ:11.97(1H,s,H-9),8.52(1H,d,J=4.9 Hz,H-3),8.46(1H,d,J= 4.9 Hz,H-4),8.32(1H,d,J=7.9 Hz,H-5),7.81(1H,d,J=8.2 Hz,H-8),7.61(1H,m,H-7),7.32(1H,m,H-6),3.53(1H,d,J=6.8 Hz,H-3′),2.70(1H,d,J=6.8 Hz,H=2′);13C NMR (100 MHz,DMSO-d6)δ:201.8(C-4′),173.8(C-1′),141.8(C-1),137.8(C-3),135.7(C-8a),133.9(C-9a),131.2(C-4b),129.3(C-6),122.2(C-5),120.6(C-4a),120.3(C-7),119.8(C-4),113.5(C-8),31.0(C-3′),29.4(C-2′)。该化合物的碳谱和氢谱数据与文献[20]报道数据一致,故鉴定该化合物为4-(9H-β-carbolin-1-yl)-4-oxobutyric acid。
化合物7淡黄色粉末;mp.127~129 ℃;UVλmax= 236,253,274,292,365,378 nm;EI-MS:m/z234 [M]+;1H NMR(400 MHz,CDCl3)δ:9.38(1H,s,H-9),8.46(1H,d,J= 5.2 Hz,H-3),7.89(1H,d,J= 5.2 Hz,H-4),8.15(1H,d,J= 7.7 Hz,H-5),7.58(1H,m,H-6),7.35(1H,m,H-7),7.33(1H,overlapped,H-8),6.71(1H,dd,J= 3.4,1.7 Hz,H-3′),7.64(1H,m,H-4′),7.74(1H,m,H-5′);13C NMR(100 MHz,CDCl3)δ:154.6(C-2′),142.8(C-5′),140.4(C-1),138.9(C-3),133.3(C-8a),131.4(C-4b),130.3(C-9a),128.6(C-6),121.7(C-5),121.2(C-4a),120.1(C-7),113.7(C-4),112.4(C-3′),111.5(C-4′),108.7(C-8)。经查阅文献[21,22],鉴定该化合物7为1-(furan-2-yl)-9H-pyrido[3,4-b]indole。
化合物8淡黄色粉末;1H NMR(400 MHz,DMSO-d6)δ:10.80(1H,s,-NH),7.52(1H,d,J=7.7 Hz,H-4),7.33(1H,d,J=8.1 Hz,H-7),7.14(1H,d,J=2.2 Hz,H-2),7.06(1H,m,H-6),6.97(1H,m,H-5),3.65(2H,t,J=7.2 Hz,H-2′),2.85(2H,t,J=7.2 Hz,H-1′);13C NMR(100 MHz,DMSO-d6)δ:136.6(C-7a),127.7(C-3a),123.2(C-2),121.2(C-6),118.8(C-5),118.6(C-4),112.0(C-7),111.7(C-3),62.2(C-2′),29.3(C-1′)。通过分析化合物8的1H NMR谱和13C NMR谱,该化合物为一个吲哚类生物碱,经查阅文献[23],鉴定该化合物为indole-3-ethanol。
化合物9淡黄色粉末;分子式:C10H11NO,mp.163~165 ℃;1H NMR(400 MHz,DMSO-d6)δ:10.90(1H,s,-NH),7.51(1H,d,J=7.7 Hz,H-4),7.35(1H,d,J=8.0 Hz,H-7),7.23(1H,d,J=2.2 Hz,H-2),7.09(1H,m,H-6),6.99(1H,m,H-5),3.64(2H,s,H-1′);13C NMR(100 MHz,DMSO-d6)δ:173.8(C-2′),136.6(C-7a),127.9(C-3a),123.2(C-2),121.2(C-6),118.8(C-5),118.6(C-4),112.0(C-7),108.3(C-3),31.6(C-1′)。通过分析化合物9的1H NMR谱和13C NMR谱,该化合物为一个吲哚类生物碱,经查阅文献[24],鉴定该化合物为indole-3-acetic acid。
化合物10淡黄色粉末;分子式:C9H7NO,EI-MS:m/z145 [M]+。1H NMR(400 MHz,DMSO-d6)δ:9.94(1H,s,-NH),8.29(1H,s,H-2),8.10(1H,d,J=7.1 Hz,H-4),7.53(1H,d,J=7.6 Hz,H-7),7.27(1H,td,J=7.5,1.3 Hz,H-6),7.23(1H,td,J=7.5,1.3 Hz,H-5);13C NMR(100 MHz,DMSO-d6)δ:185.4(C-1′),139.0(C-7a),137.7(C-2),124.7(C-3a),121.3(C-4),118.6(C-3),123.9(C-6),122.5(C-5),112.9(C-7)。通过分析化合物10的1H NMR谱和13C NMR谱,该化合物为一个吲哚类生物碱,经查阅文献[25],鉴定该化合物为indole-3-carboxaldehyde。
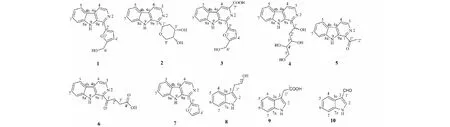
图2 化合物1~10的结构Fig.2 The chemical structures of compounds 1-10
3.2 黄嘌呤氧化酶抑制实验结果
对化合物1~10进行抑酶活性筛选,通过测试酶反应后与显色剂生成的紫色化合物吸光度的大小可间接反应黄嘌呤氧化酶的活性,紫色化合物吸光度越大,酶活性越强。不同浓度的别嘌呤醇作为阳性药对黄嘌呤氧化酶抑制作用的测试结果如表1所示,其半数抑制浓度为1.61 mM,与文献[12,26]报道数据较一致,具有一定重复性和参考价值。10个化合物对黄嘌呤氧化酶抑制作用的测定结果如表2所示,其中7个化合物具有不同程度的抑制作用,化合物2、3和6对黄嘌呤氧化酶的半数抑制浓度最小,分别为0.90、0.98和0.68 mM,且明显好于别嘌呤醇。与川芎哚(化合物1)(IC50=2.84 mM)的活性相比,4个川芎哚类似物(化合物2~4、6)的抑制作用更佳。
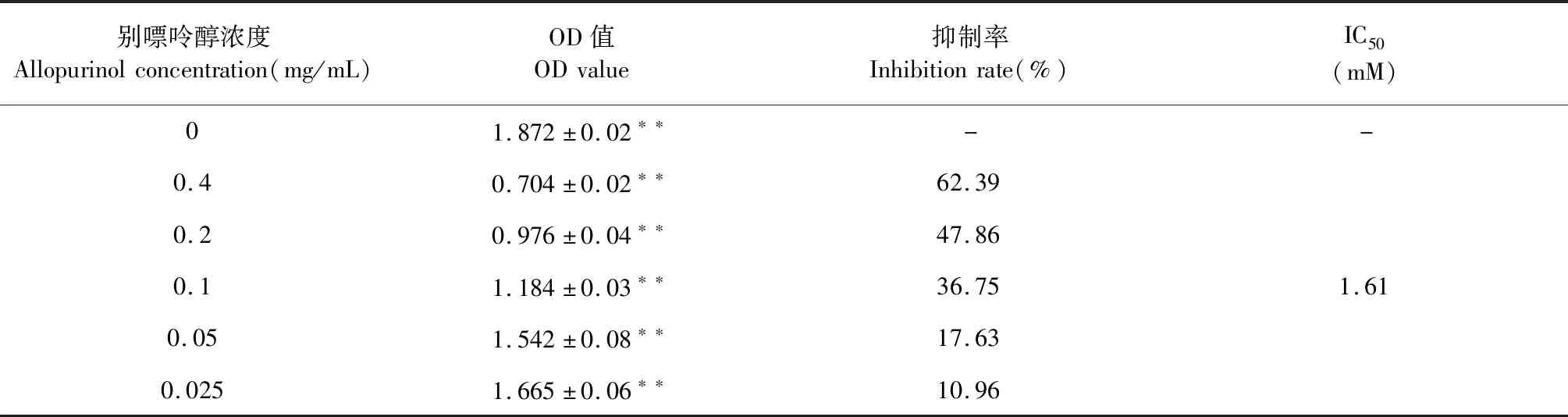
表1 别嘌呤醇对黄嘌呤氧化酶的抑制作用结果(n=3)
4 讨论与结论
微生物来源的天然产物与植物来源相比具有明显的优势,具有生长周期短、可代谢调控、可规模化生产等特点。本课题菌株虽然可产生川芎哚,但在发酵产物中含量很低。关于川芎哚的生物合成途径研究也未见报道,如何通过菌株的代谢调控,提高川芎哚或其类似物的产量,是本课题继续开展的关键所在。查阅文献发现,川芎哚化学合成中最重要的一步是L-色氨酸和5-乙酰氧甲基糠醛通过Pictet-Spengler(P-S)反应的环化,且P-S反应酶(Pictet-Spenglerase)是β-咔啉类生物碱合成的关键酶[27]。所以,本课题通过对真菌进行前体介导从而完成次级代谢产物调控,对提取物浸膏进行LC-MS分析,发现其中含有大量生物碱,分子量在m/z200~400之间,符合川芎哚类似物的分子量推测,因此,可以推测,L-色氨酸前体的加入,激活了β-咔啉类似物的沉默基因簇的表达,合成了大量的生物碱。本课题通过研究从中获得了10个生物碱类化合物,以上化合物均为首次从该属菌株中分离得到,其中7个β-咔啉生物碱,6个川芎哚类似物,有针对性的提高了目的产物的产量和种类;对10个生物碱进行了黄嘌呤氧化酶抑制活性测试,结果显示7个化合物具有明显的抑制活性,IC50值的范围在0.68~9.30 mM之间,化合物2、3和6的抑制作用效果明显,且优于阳性药。4个川芎哚类似物(化合物2~4、6)与川芎哚(化合物1)的活性相比,抑制作用更佳。Li等[10]从酱油中也分离得到化合物3,生物活性筛选结果表明,化合物3具有较强的黄嘌呤氧化酶抑制活性(IC50= 0.51 ± 0.05 mM),与本文实验数据较一致,他们根据荧光猝灭和分子对接实验,发现化合物3能通过疏水作用力和氢键进入黄嘌呤氧化酶催化中心与氨基酸Lys1045、Gln1194和Arg912相互作用产生抑制活性;而且该化合物的ADME(吸收、分布、代谢、排泄)药物体内过程结果表明,其在体内具有良好的口服生物利用度;此外,该课题组[28]还发现化合物3有较强的抗HIV活性(EC50=2.36 μM,TI=12.1),经过结构修饰得到flazinamide后,发现其显示出很强的抗HIV-1活性(EC50=0.38μM,TI=312.0),现已进入临床前各项研究工作。另外有研究表明[29],在四氢-β-咔啉的3位引入羧酸侧链,可提高其生物溶解度,使其具有更高的抗血栓活性及抗血小板聚集活性,而化合物3即为川芎哚的3位羧酸化产物。结合以上文献报道,对化合物1~10的初步构效关系分析,可以看出,β-咔啉生物碱的3位羧酸化(化合物3),及1位侧链的羟基化(化合物2)或羧基化(化合物6),均能够有效提高黄嘌呤氧化酶抑制活性。近年来,从动物实验到人体研究都证实了黄嘌呤氧化酶与心血管疾病密切相关,黄嘌呤氧化酶抑制剂可应用于多种心血管疾病的治疗。本文的研究结果对于川芎哚及其类似物的产量和种类提供了思路,并为开发出具有优良利用价值的黄嘌呤氧化酶抑制剂提供了参考依据。
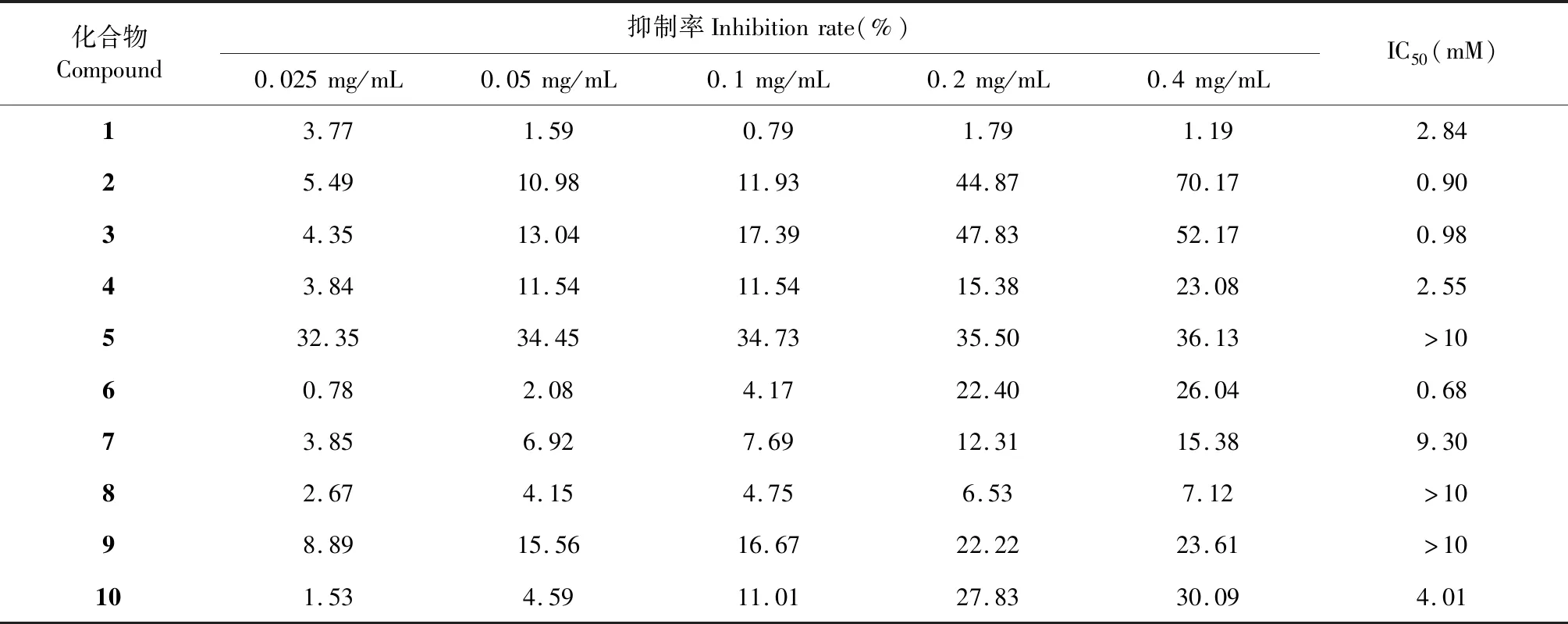
表2 不同浓度化合物1~10对黄嘌呤氧化酶的抑制率(n=3)
