中年男性,发作性头晕2年,右侧肢体无力2天
——CADASIL 2型
2021-01-30朱敏周美鸿洪道俊
朱敏 周美鸿 洪道俊
1 资料
患者男,45岁,因“发作性头晕2年,右侧肢体无力2 d”于2020年5月20日就诊于我院神经内科。
患者(III3,见图1)于 2018年2月出现头晕,伴行走不稳感,症状持续数天逐步缓解,当时无视物旋转,无恶心呕吐,无耳鸣及听力下降,无肢体无力,未引起重视,未至医院就诊。2018年5月再次出现头晕发作,伴右侧肢体麻木,无肢体无力,无视物成双,无吞咽困难,无饮食呛咳,无言语含糊等症状。行头颅MRI检查提示左侧脑室旁急性腔隙性梗死病灶,伴白质病变。经过治疗后患者症状改善,未遗留明显后遗症,之后一直服用拜阿司匹林和他汀类药物治疗。2019年7月再次出现头晕,无其他伴随症状,复查头颅MRI提示右侧脑桥急性腔隙性脑梗死。此次入院患者2 d前突发右侧肢体无力,表现为右手抬举费力,右下肢走路拖拽,伴言语含糊不清,偶有饮水呛咳,无肢体麻木,无头晕头痛,无视野缺损。病程中患者无腰痛,无头痛,大小便正常。患者长期睡眠不好,情绪较焦虑,饮食一般但食欲不佳,体重无明显变化。
既往史及家族史:患者银屑病史10余年,使用生物制剂,但具体药物不详,症状控制尚可。否认高血压和糖尿病史等慢性病史,否认吸烟及饮酒史,否认关节疼痛。母亲(II3)50岁发生脑出血,52岁因脑出血死亡,也存在秃发情况,否认高血压及血管畸形病史;弟弟(III4)43岁,40岁发现额顶秃发,无腰痛,42岁发生1次脑梗死,头颅MRI显示多发陈旧性腔梗伴脑白质病变,现服用波立维、阿托伐他汀及盐酸美金刚,无高血压及糖尿病病史,无吸烟及饮酒病史;大舅舅(II2)20岁时出现精神障碍,60余岁死亡,原因不详;三舅舅(II5)20岁因脑肿瘤死亡(图 1A)。
体格检查:体温36.7℃,脉搏90次/min,呼吸15次/min,血压122 mmHg/80 mmHg。双肺呼吸音浊,闻及少量湿啰音,心律齐,各瓣膜区听诊未闻及杂音。患者额顶头发稀疏(图1B),约40岁左右明显。神志清楚,言语稍含糊。简易智能精神状态检查量表评分(MMSE)27分,蒙特利尔认知评估量表(MoCA)23分。双侧瞳孔等大正圆,直径约3.0 mm,直接及间接对光反射灵敏。双眼睑闭合力可,双侧眼球向各个方向运动正常,未及复视和眼震。角膜反射正常,双侧面部痛温觉无减退。双侧颞肌、咀嚼肌对称,咬肌力弱。双侧额纹,右侧鼻唇沟变浅。双侧听力粗测正常。悬雍垂居中,双侧软腭上抬欠佳,咽反射存在。双侧转颈、耸肩力弱。伸舌稍偏右,无舌肌萎缩及震颤。颈屈肌力5级。四肢未见明显肌萎缩,四肢肌张力正常,右上肢肌力4+级,右下肢肌力5-级,左侧肢体肌力5级,双侧肢体及躯干痛温觉、位置觉、振动觉及运动觉正常。四肢腱反射活跃。双侧Hoffman′s征阳性,双侧 Babinski′s 征、Oppenheim′s 征、Gordon′s 征、Chaddock′s征均阴性,掌颌反射和吸吮反射阳性。脑膜刺激征阴性。自主神经功能检查正常。
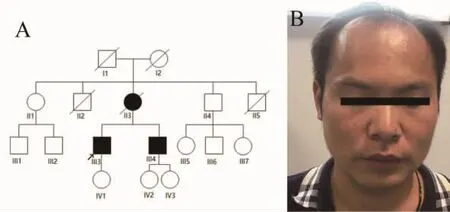
图1 家系图和先证者头发改变 家系图(A)箭头所示为先证者,家系内有母亲和弟弟有卒中病史。先证者前额顶头发变少,稀疏(B)。
辅助检查:血、尿、便常规正常,肝肾功能、血脂、电解质、肌酶谱、同型半胱氨酸均正常,甲状腺功能正常。梅毒抗体、抗HIV抗体阴性。糖化血红蛋白6.4%(4.5%~6.3%),空腹血糖 6.27 mmol/L(3.9~6.1 mmol/L),OGTT 试验餐后2 h 血糖 13.84 mmol/L。 血沉 42 mm/h(1~20 mm/h),C反应蛋白 42.3 mg/L(0~8 mg/L)。颈部血管彩超:双侧劲动脉、椎动脉及锁骨下动脉未见异常。心脏彩超正常。头颅MRI(图2A-E)示双侧基底节区新发脑梗塞,皮层下及侧脑室旁脑白质病变,并陈旧性脑梗塞病灶,脑干也可见微出血及陈旧性病灶;磁敏感示基底节区、皮层及皮层下多发微出血。头颈部CTA示未见明显异常(2F)。
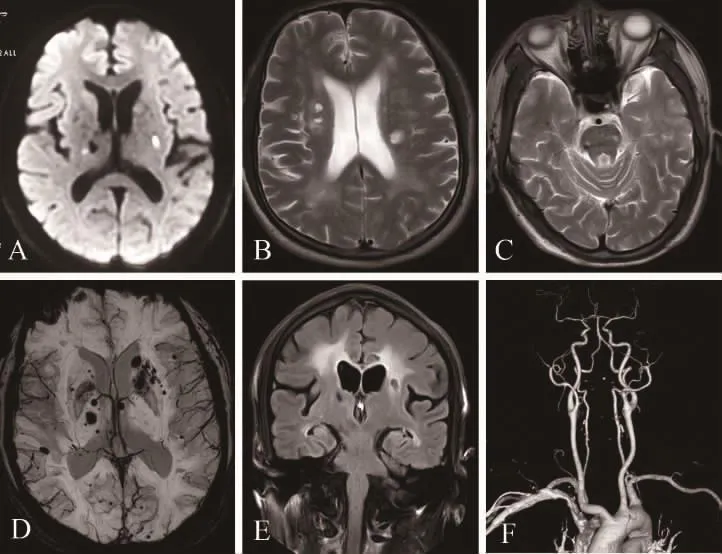
图2 先证者头颅影像检查 弥散像示双侧基底节区新发腔隙性脑梗死灶(A),T2像示皮层下及侧脑室旁脑白质病变,并陈旧性脑梗塞病灶(B),脑干可见微出血及陈旧性病灶(C),磁敏感示基底节区、皮层及皮层下多发微出血(D),压水像示侧脑室旁脑白质改变(E),头颈部 CTA 示正常(F)。
基因检查:二代测序显示高温需求丝氨酸肽酶1(HTRA1)基因上存在一个杂合子突变 c.1061C>T(p.A354V),家系验证其父亲为野生型,母亲因死亡未验证。其弟弟也携带该突变,患者女儿18岁,也携带该突变点,目前未出现症状,需进一步随访观察,一代测序也证实该突变(图3)。该位点位于丝氨酸蛋白酶结构域,在物种之间为高度保守。致病性预测均提示致病包括:PolyPhen-2为可能有害,MutationTaste为致病,PROVEAN为有害。虽然未得到患者母亲的验证,但结合临床推断母亲携带该致病基因,因此认为该突变与常染色体显性遗传性脑动脉病伴皮质下梗死脑白质病2型(CADASIL 2型;OMIM:616779)密切相关。
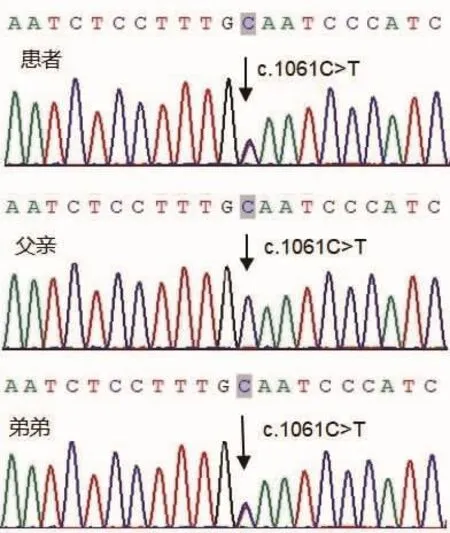
图3 基因突变分析 先证者HTRA1基因上存在一个杂合子突变c.1061C>T(p.A354V),与患者有类似症状的弟弟有同样突变,其父亲为野生型。
诊断明确后,给予该患者西洛他唑50 mg,2次/d;盐酸多奈哌齐,5 mg,1 次/d;草酸艾司西酞普兰,20 mg,1 次/d;阿卡波糖,50 mg,3次/d。1个月后复诊,患者自述行动能力基本恢复正常,睡眠尚可,精神状态正常。MMSE评分28分,MoCA评分25分。
2 讨论
脑小血管病(cerebral small vessel disease,CSVD)指累及大脑小动脉、穿支动脉、毛细血管和小静脉的一组疾病。可分为散发性和遗传性,目前有5%的脑小血管病可以归因于单基因遗传,主要包括NOTCH3基因突变导致的伴有皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(CADASIL 1型)及HTRA1基因突变导致的伴有皮质下梗死和白质脑病的常染色体隐性遗传性脑动脉病(CARASIL)[1]。这其中CARASIL更为少见,临床主要表现为青年时期反复发作的脑卒中、白质脑病,以及反复腰痛、脱发等症状,无高血压、糖尿病等脑血管病危险因素。其发病与10号染色体的 HTRA1基因突变导致转化生长因子-β家族转导的信号异常升高,从而使血管容易形成纤维化有关[2],以纯合突变为主,少数表现为复合杂合突变[3]。但最近几年发现杂合子HTRA1突变与常染色体显性遗传性脑小血管疾病有关,即 CADASIL 2 型[4-6]。
HTRA1基因杂合突变导致的CADASIL 2型是一种非常罕见的脑血管病。自2015年VERDURA等[5]发现杂合子HTRA1基因突变与常染色体显性遗传性脑血小血管有关以来,目前大约50例患者被报告[7],主要分布在亚洲[5]包括中国[1,8-10]、日本[4]、以及欧洲[5]包括意大利[6,11-12],个别在欧洲的德国[13]、希腊[14]和非洲[15]。研究表明在NOTCH3突变阴性的遗传性脑血管病家系中,中国台湾有5.61%的携带杂合 HTRA1 突变[1],日本为4.98%[4],意大利 3.52%[6]。上述数据表明HTRA1突变导致的CADASIL 2型较HTRA1突变导致的CARASIL更常见,到目前为止,我们中心仅诊断1例CARASIL患者,但已经诊断了2个CADASIL 2型家系,提示CADASIL 2型可能是更常见的类型,也提示筛查HTRA1基因在家族性脑血管病的重要性。
CADASIL 2型的主要临床表现包括神经系统症状和神经系统外症状。神经系统症状包括反复卒中发作(腔隙性脑梗塞、短暂性脑缺血发作、脑出血)、步态异常、认知功能障碍、情绪改变,以及球麻痹等。神经系统外表现有腰痛、脊椎病、秃头或者头发稀疏。该例患者神经系统症状表现出典型的神经系统损害症候群,同时家系内多例患者出现秃发现象,因此临床表型上和文献报道的非常相似。这些症状与CARASIL症状也存在较高的相似性,但两者之间存在一些可以鉴别的特点,主要表现在以下几个方面:①CADASIL 2型男性多见(男性为 76.1%),而 CARASIL男女比例均衡(男性为42.9%);②CADASIL 2型患者神经系统症状平均发病年龄较CARASIL晚,两者平均发病年龄分别为54.1岁和29.5岁,文献报道CADASIL 2型的最大发病年龄可达77岁,如果家族史不明确,容易考虑是其他原因导致的脑卒中[10];③CADASIL 2型患者神经系统以外的症状较CARASIL少见,文献报告仅13.2%的CADASIL 2型患者出现秃头,而在CARASIL患者中达85.7%;脊椎病或腰痛在CADASIL 2型中有62%出现,而几乎所有的CARASIL都存在脊柱病变[7]。
CADASIL 2型的头颅影像学主要表现为侧脑室旁及深部的脑白质病变,基底节区、胼胝体、脑干等部位的腔隙性脑梗死或微出血病灶也是常见的影像学特征,脑白质病变通常不累及 U 型肌纤维[6,8,11],也罕见到 CADASIL 1 型患者颞极的白质病变[1,12]。该患者头颅MRI在T2像显示在脑干、侧脑室旁及基底节区多发腔隙梗死病灶并白质病变,单纯从影像学特点很难和其他类型的脑小动脉病变鉴别,然而该家系患者的母亲死于明确的脑出血,先证者本身的头颅MRI显示存在显著的微出血病灶,提示部分CADASIL2患者可以类似于CADASIL 1型一样出现严重的脑出血。
目前有2例CADASIL 2型进行了脑血管病理学检查,颅内血管病理显示软脑膜动脉、穿通动脉以及小动脉内侧平滑肌细胞大量丧失和纤维化、内膜增生、弹性层断裂[4,16],电子显微镜示在弹性层的外层可见电子致密物沉积,但没有发现CADASIL 1型特征性的平滑肌细胞表面嗜锇颗粒沉积[16]。
导致CADASIL 2型的HTRA1突变包括错义突变、无意义突变、缺失突变,突变位置主要位于LD(283-291位氨基酸)、L3(301-304位氨基酸)和 Kazal-like与蛋白酶结合区。LD和L3结构域是通过分子间联系实现HTRA1蛋白酶活性所需的必须结构域,杂合突变HTRA1可能通过负显性作用导致HTRA1活化级联反应受损或者不能形成稳定的三聚体,最后导致蛋白酶活性下降而出现临床症状[4]。导致CARASIL的突变位点不位于上述结构域,因此CARASIL相关突变通常和HTRA1双突变导致酶活性的下降有直接关系[7]。不同程度的HTRA1蛋白酶活性下降导致的临床严重程度不同[1],而同一家族中也可以出现不同的临床症状及发病年龄不同[12],这提示在基因突变基础上,外界因素也参与了整个发病过程。该家系基因检查发现在HTRA1基因上有一杂合变异,该变异在正常人数据库中没有报道,但是在该家系中出现共分离现象,而且受累家系成员没有明确的脑血管病危险因素,且该突变位于丝氨酸蛋白酶结构域,因此该突变被认为是CADASIL 2型的致病突变。
脑小动脉病变由于其病理生理的机制导致在药物的二级预防方面一直缺少有效的手段,特别对遗传性脑小动脉病变方面更是缺少循证医学研究。西洛他唑对于伴有多发微出血的脑小血管病患者可能有减少脑出血的风险,因此抗血小板治疗时作为首先推荐。此外,CADASIL 2型患者常常伴随认知障碍和情感障碍,相应的对症治疗对患者也有帮助。
3 点评
由于遗传学和影像学的进展,脑小动脉病变已经成为血管病研究的一个热点领域。依据脑小动脉病有提示价值影像学特征:脑白质病变、腔隙性梗死灶、微出血病灶、Virchow间隙增大,结合较年轻的发病年龄,以及不显著的脑血管病危险因素,临床医师要时刻警惕遗传性脑小动脉病的可能。目前遗传性脑小动脉病的典型代表包括NOTCH3基因导致的CADASIL 1型;HTRA1基因突变导致CADASIL 2型和CARASIL;GLA基因突变导致的Fabry病;COL4A1和COL4A2基因突变导致的COL4A动脉病。这其中最常见的当为NOTCH3相关的CADASIL 1型,但是令人印象最深刻的却是HTRA1基因的临床表型,在HTRA1研究的早期一直是在近亲结婚的家庭中发现纯合子突变导致CARASIL,甚至在很长的一段时间内,近亲结婚成为CARASIL的诊断要点之一,但不成想HTRA1特定结构域的突变可以导致显性遗传的CADASIL 2型。类似现象也见于多种其他遗传病中,例如Calpain-3基因突变长期被认为仅导致常染色体隐性肢带型肌营养不良1型(LGMDR1),但是近年发现Calpain-3基因的单个杂合突变可以导致常染色体显性遗传肢带型肌营养不良4型(LGMDD4)[17]。这种基因表型和临床表型之间扩展现象,随着基因测序的可获得性增高,越来越成为可能发现,也提醒医学研究者要有原创性的胆识和敏锐度。
