滇黄精转录组测序及类黄酮合成相关基因的分析
2020-12-29肖韵铮韩世明秦昭李春奇
肖韵铮, 韩世明, 秦昭, 李春奇
(1.河南农业大学生命科学学院,河南 郑州 450002;2.六盘水师范学院生物科学与技术学院,贵州 六盘水 553000)
黄精(Polygonati rhizoma)是著名的药食两用植物,包括黄精(PolygonatumsiricumRed.)、滇黄精(PolygonatumkingianumColl.et Hemsl.)等品种。滇黄精(PolygonatumkingianumColl.et Hemsl)为百合科黄精属多年生草本植物,主要分布于中国西南地区,主产于云南省,以根茎入药,是中国常用的中草药[1]。滇黄精富含黄酮类化合物(flavonoids)。黄酮类化合物又称类黄酮,是植物中重要的次生代谢产物,具有抗衰老、降血糖、改善血液循环等保健功能,具有很高的经济价值和药用价值[2-3]。目前,滇黄精和其次生代谢产物合成机理鲜有报道,尤其对滇黄精类黄酮合成的分子机理研究较少。因此,本试验以滇黄精为研究对象进行转录水平分析,并对影响滇黄精类黄酮含量的关键基因进行了系统研究,为解析类黄酮生物合成分子机制奠定基础。
转录组测序(RNA-Seq)技术是利用高通量测序技术对生物进行转录组分析,目前已广泛应用于生物基因组基因表达的研究[4-7],包括人参[8]、柴胡[9]以及甘草[10]等药用植物已通过该技术进行了转录组测序。RNA-Seq技术不仅可以用于检测和分析有基因组序列的生物体中的转录本,还可以用于组装短片段,在无参考基因组的生物体中进行基因发现和基因表达分析[11]。鉴于此,本研究以滇黄精根、茎和叶为对象进行转录组测序,通过数据分析挖掘与类黄酮生物合成途径相关的功能基因,为滇黄精功能基因鉴定和遗传改良奠定研究基础。
1 材料和方法
1.1 试验材料
滇黄精植株由六盘水师范学院提供。选择9株生长状态良好且长势相同的6个月盆栽苗。每3株为1组,共3组。第1组取根、茎和叶分别放入 3 个离心管中,其余组取材时选取发育阶段相似、部位相同的根、茎和叶,共计9个样本,3次生物学重复。取样后立即置于液氮速冻保存于-80 ℃备用。
1.2 RNA提取、文库的制备和测序
从滇黄精器官样品(根、茎和叶)中提取总RNA,RNA提取使用植物总RNA提取试剂盒(Quick RNA isolation Kit,北京华越洋生物科技有限公司)。使用微量紫外分光光度计(Thermo Scientific, NANO, DROP2000)对总RNA进行定量,并通过测量A260/A280和A260/A230检测总RNA的纯度,将纯化的RNA溶解在RNasefree水中并储存在-80 ℃。测得样品质量浓度不小于400 mg·L-1,OD260/OD280=1.8~2.2,28S∶18S≥1.5∶1.0。将总RNA反转录成cDNA,并通过PCR扩增富集得到最终的cDNA文库,完成文库制备。库检合格后,使用Illumina HiSeq 2500测序平台(北京百迈客生物科技有限公司)进行测序。
1.3 质量控制、序列组装和功能注释
采用Illumina HiSeq 2500高通量测序平台对cDNA文库进行测序,去除reads中的测序接头、引物序列以及低质量reads(质量值Q<30),对于得到的高质量reads(clean reads)用Trinity软件进行组装。从获得的基因功能信息中,将组装得到的滇黄精的转录本数据与公共数据库进行BLAST比对,并使用HMMER软件与Pfam数据库比对获得注释信息。公共数据库为非冗余蛋白质数据库(Nr)(ftp://ftp.ncbi.nih.gov/blast/db/)、蛋白质家族(Pfam)(http://pfam.xfam.org/)、直系同源蛋白质簇(KOG/COG/eggNOG)(http://www.ncbi.nlm.nih.gov/KOG/; http://www.ncbi.nlm.nih.gov/COG/; http://eggnogdb.embl.de/)、Swiss-Prot 蛋白序列数据库(Swiss-Prot)(http://www.uniprot.org/)、基因本体数据库(GO)(http://www.geneontology.org/)和京都基因与基因组百科全书(KEGG)(http://www.genome.jp/kegg/)。BLAST参数E-value≤10-5,HMMER软件参数E-value≤10-10,通过序列相似性进行功能注释。
1.4 差异表达基因的筛选
采用Bowtie软件[12]将测序获得的 reads与unigenes库比对,并使用 RSEM软件[13]根据比对结果对表达量水平进行估计。使用每百万reads中比对到某一基因每千碱基长度的reads数目(fragments per kilobase of transcript per million mapped reads, FPKM)表示对应unigenes的表达丰度。然后用DESeq方法[14]对样品进行差异表达分析,错误发现率(false discovery rate,FDR)<0.05且倍数差异(fold change,FC)>2作为筛选标准。GO功能富集采用topGO软件(http://www.bioconductor.org/packages/release/bioc/html/topGO.html)进行分析,使用KOBAS2.0(http://kobas.cbi.pku.edu.cn/help.do)对差异表达基因(differentially expressed genes, DEGs)进行KEGG富集分析。
1.5 类黄酮含量的测定
以芦丁标准品(生工生物工程上海股份有限公司,分析纯)来配制芦丁标准溶液(质量分数80%乙醇为溶剂),以质量分数80%乙醇作为对照,利用紫外分光光度计测定510 nm 处的吸光值。以芦丁质量浓度为横坐标,吸光度值为纵坐标,绘制出标准曲线。
测定滇黄精不同器官中类黄酮的含量,参照刘清华等[15]的方法稍作修改。称取滇黄精根、茎和叶样品各0.2 g,按照料液比m(样品)∶V(质量分数80%乙醇)=1∶15分别置于10 mL试管中,超声提取50 min,后10 000 r·min-1离心10 min,取1 mL上清液用质量分数80%乙醇溶液稀释到5 mL,吸取2 mL定容至10 mL,测定其在510 nm处吸光值,以质量分数80%乙醇作为对照,查询标准曲线计算提取液中类黄酮质量浓度,再按下式计算不同器官中类黄酮含量:
M=(c×v×d)/m
(1)
式中:M表示类黄酮含量;c表示提取液中类黄酮质量浓度;v表示待测提取液总体积;d表示总稀释倍数;m表示滇黄精样品质量。
1.6 差异表达基因的qRT-PCR
总RNA提取使用植物总RNA提取试剂盒(Quick RNA isolation Kit,北京华越洋生物科技有限公司)。使用FSQ-101反转录试剂盒(东洋纺(上海)生物科技有限公司)将mRNA反转录为cDNA。依据转录组测序结果,筛选得到10个与类黄酮合成相关的差异基因,通过实时荧光定量PCR仪(LC480,瑞士罗氏集团)以及SYBR qPCR Mix试剂盒(北京艾德莱生物科技有限公司)进行qRT-PCR实验。qPCR的引物由NCBI/Primer-BLAST界面进行设计,引物由生工生物工程(上海)股份有限公司合成,如表1所示。引物序列长度范围18~25 bp,基因扩增产物长度限制100~350 bp。以18S rRNA为内参基因,上游引物为CTTGCGACCTGGAGTTATTGAT,下游引物为CTGCAACACTGCTCTTGCTC[16]。PCR反应体系和反应程序参照崔雯等[17]方法。
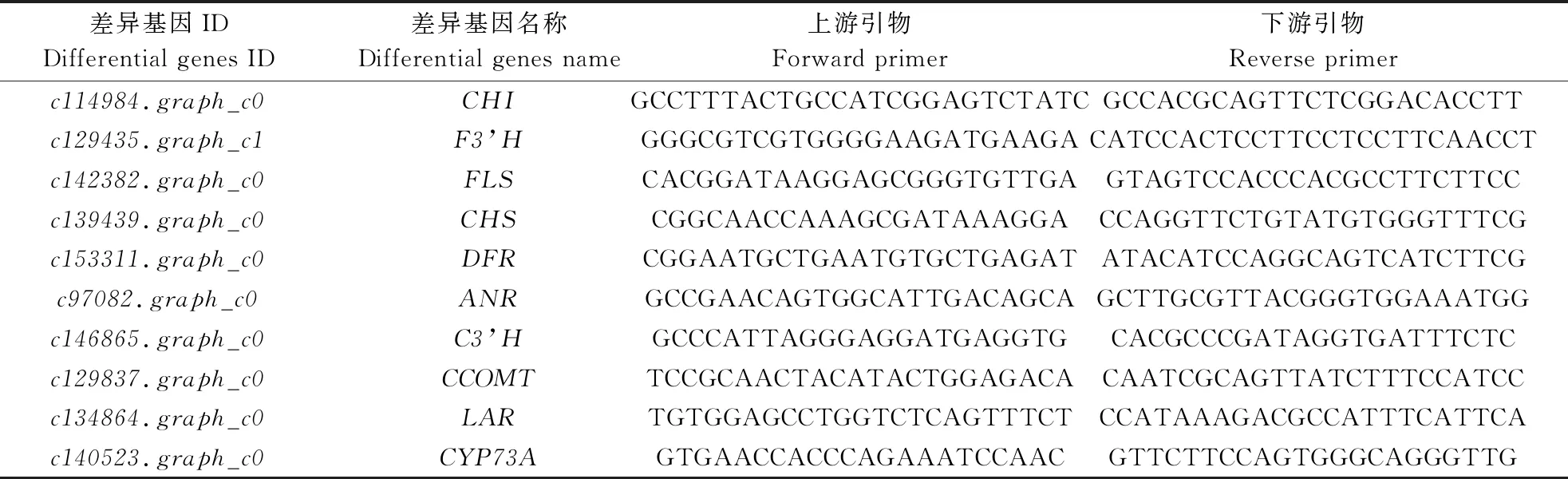
表1 荧光定量PCR引物
2 结果与分析
2.1 滇黄精转录组数据组装分析结果
采用Illumina HiSeq 2500测序平台技术得到滇黄精转录组数据。对获得的原始数据进行统计,完成滇黄精样品的转录组测序,去除raw data中的接头序列及低质量reads获得高质量的clean data 40.92 Gb,各样品clean data均达到5.93 Gb,Q30碱基比例在91.94%以上,通过组装后共产生了390 456条转录本,平均长度为770.38 bp,转录本N50长度为1 350 bp。对转录本序列进一步组装拼接,共获得了219 769条unigenes序列,平均长度为502.31 bp,N50长度为627 bp。其中,转录本在200~300 bp之间有139 323条,300~500 bp 之间有83 204条,大于2 000 bp的有32 976条,如图1所示。
2.2 滇黄精unigenes序列功能注释
将获得的unigenes进行基因功能注释以得到全面的基因功能信息,数据库分别为Nr、Pfam、KOG/COG、Swiss-Prot、eggNOG、KEGG以及GO。经过BLAST程序对比,在219 769条滇黄精unigenes中,共有81 853条unigenes被注释到,覆盖率为37.2%。如表2所示,以数据库Nr注释成功率最高(34.7%),其次为eggNOG(30.5%)、Pfam(20.7%)、KOG(19.7%)、GO(18.0%)、Swiss-Prot(16.1%)、KEGG(11.9%)和COG(11.2%)。

图1 滇黄精转录本数据和unigenes的长度分布情况
2.3 滇黄精unigenes的GO和KOG功能分类
2.3.1 滇黄精unigenes的GO分类 根据unigenes的GO功能分析,滇黄精unigenes的GO注释结果如图2所示。在219 769条unigenes中,共有39 479条unigenes(18.0%)被注释到GO条目中,并被分为3个主要类别,即生物过程(99 866条)、分子功能(48 515条)和细胞组分(78 215条)。在20个生物过程中,unigenes最多的为参与代谢过程(28 018条)和细胞过程(23 119条),其次是单一有机体过程(17 934条);在18个细胞组分中,unigenes最多的是细胞(18 352条)和细胞组分(18 352条),其次是细胞器(14 245条)和细胞膜(8 512条);在16个分子功能中,unigenes最多的是催化活性(21 212条)和绑定功能(19 999条)。
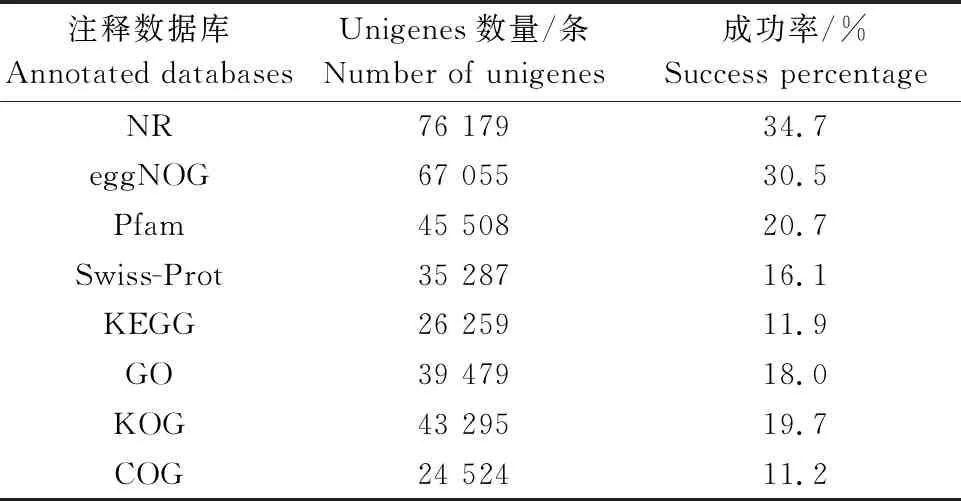
表2 Unigenes注释的成功率统计

注: 1.胞外区; 2.胶原三聚体;3.细胞; 4.拟核; 5.细胞膜; 6.病毒粒子;7.细胞连接;8.胞外基质; 9.膜内腔;10.大分子复合物; 11.细胞器 12.细胞外基质部分;13.胞外区组件;14.细胞器组件;15.病毒粒子部分;16.细胞膜组件;17.细胞组件;18.共质体;19.蛋白质结合转录因子活性;20.核酸结合转录因子活性;21.催化活性; 22.受体活性; 23.鸟苷酸交换因子活性;24.结构分子活性;25.转运器活性;26.绑定; 27.电子载体活性; 28.抗氧化活性; 29.金属伴侣活性 30.酶调节剂活性; 31.蛋白标签 32.翻译调节器活性;33.营养储层活动; 34.分子传感器活性;35.生殖;36.细胞杀伤;37.免疫系统进程 ;38.代谢进程;39.细胞进程; 40.生殖进程;41.生物黏附 ; 42.信号;43.多细胞有机体进程;44.发育进程 ;45.生长; 46.运动力; 47.单一有机体进程;48.生物相;49.节律进程;50.应激反应; 51.定位;52.多有机体进程;53.生物调节; 54.细胞成分组织或生物合成。
2.3.2 滇黄精unigenes的KOG分类 将组装得到的unigenes在KOG数据库中进行比对,共有43 295条unigenes具有明显的序列相似性且被注释到25个分类中,如图3所示。在滇黄精器官中,一般功能预测类的unigenes最多为10 244条,其次是翻译后修饰为4 904条;而细胞运动是最少的类别为44条。

注: A: RNA加工和修饰; B: 染色质结构与动力; C: 能量的产生及转换; D: 细胞周期控制, 细胞分裂, 染色体分区; E: 氨基酸运输与代谢; F: 核苷酸运输与代谢; G: 碳水化合物的运输与代谢; H: 辅酶运输与代谢; I: 脂质运输与代谢; J: 翻译, 核糖体结构和生物转化; K: 转录; L: 复制, 重组和修复; M: 细胞壁, 细胞膜的发生; N: 细胞移动; O:蛋白翻译后修饰, 蛋白质转换,分子伴侣; P: 无机离子运输与代谢; Q: 次生代谢产物生物合成, 运输和分解代谢; R: 一般功能基因; S:功能未知; T: 信号转导机制; U: 细胞内运输, 分泌和膜泡运输; V: 防御机制; W: 细胞外结构; Y: 细胞核结构; Z: 细胞骨架。
2.4 滇黄精差异基因的表达分析
滇黄精3个器官中共同表达的 unigenes有20 573条,根和茎、根和叶以及茎和叶中分别有930、 256、 1 171条unigenes特异表达,如图4所示。以茎为对照,根中有2 361条DEGs,包括921条上调和1 440条下调;以叶为对照,根中有2 862条DEGs,包括1 340条上调和1 522条下调;以叶为对照,茎中有100条DEGs,包括27条上调和73条下调,如图5所示。
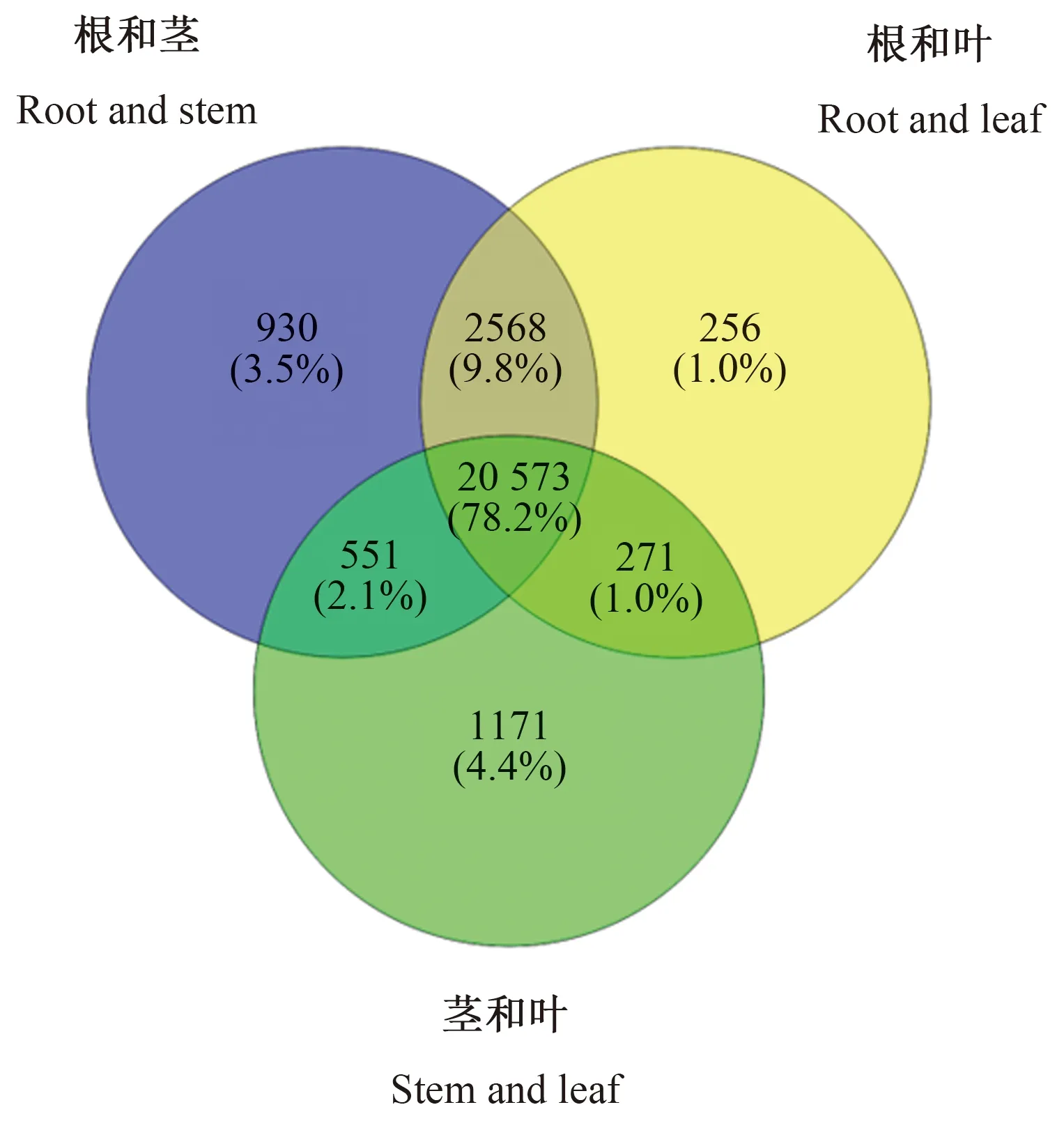
图4 滇黄精Unigenes的维恩图

图5 滇黄精差异表达基因数目
2.5 滇黄精差异基因功能注释和富集分析
在7个数据库中,本研究对差异表达基因进行功能注释,并且绘制了不同器官之间(根和茎、根和叶、茎和叶)差异表达基因的GO功能分类图,如表3和图6所示,图中横坐标表示GO的3个模块(生物学过程、 细胞组分、分子功能),纵坐标左侧表示差异表达基因数量,右侧表示所有基因数量。利用KEGG代谢途径数据库对差异表达基因KEGG的注释结果进行分类,如图7所示,图中横坐标表示的是注释到该通路下的基因数量及其与被注释上的基因总数的比例,纵坐标表示的是KEGG代谢通路的名称。根据注释结果,共有26 259条(11.9%)unigenes注释到KEGG数据库中,和通路相关的为16 574条。在根和茎中,和通路相关的差异表达基因总数为483条,分别注释到105条代谢通路;在根和叶中,和通路相关的差异表达基因总数为560条,分别注释到108条代谢通路;在茎和叶中,和通路相关的差异表达基因总数为18条,分别注释到22条通路中。富集通路主要集中在核糖体(ko03010),另外是光合作用(ko00195)、碳代谢(ko01200)、苯丙烷类生物合成(ko00940)、淀粉和蔗糖代谢(ko00500),这表明与类黄酮合成相关通路是差异基因主要富集通路之一。滇黄精unigenes代谢途径可分成5大类,涉及unigenes数量最多的前3个代谢途径分别为核糖体、碳代谢和光合作用。不同组间代谢通路比例最高,而有机系统通路比例最低。与核糖体相关的unigenes为2 254条,其次是碳代谢相关的unigenes为1 418条。对KEGG中的pathway通路进行分析,结合unigenes的注释情况,发现滇黄精根中高表达基因的功能主要与自身能量储藏代谢相关,茎和叶高表达基因功能主要与叶绿素代谢相关。
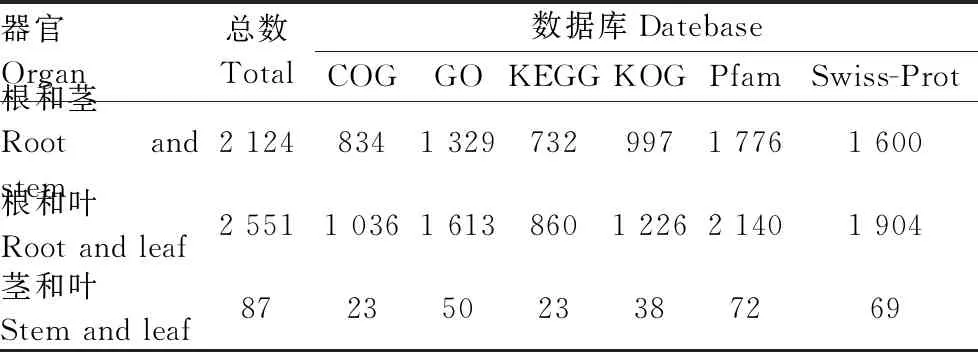
表3 滇黄精器官间注释的差异表达基因数(DEGs)

注:1.代谢过程; 2.细胞过程; 3.单一有机体进程; 4.定位;5.应激反应; 6.生物调节;7.细胞成分组织或生物合成; 8.发育过程; 9.多细胞生物过程; 10.信号;11.生殖过程; 12.多组织过程; 13.生殖; 14.生长; 15.免疫系统过程; 16.生物附着;17.生物相; 18.节律性过程;19.运动;20.细胞杀伤性;21.细胞;22.细胞组件; 23.细胞器;24.细胞膜;25.大分子复合物;26.细胞器组件;27.细胞膜组件;28.膜内腔; 29.胞外区; 30.细胞连接;31.共质体;32.拟核; 33.胞外区组件; 34.细胞外基质;35.细胞外基质部分36.病毒粒子; 37.病毒粒子组件;38.胶原三聚体; 39.催化活性;40.结合;41.转运活性; 42.结构分子活性; 43.电子载体活性;44.核酸结合转录因子活性;45.抗氧化活性;46.酶调节剂活性47.分子转导活性;48.受体活性; 49.鸟苷酸交换因子活性 50.转录因子活性,蛋白结合;51.营养库活性;52.金属伴侣活性;53.蛋白标签; 54.翻译调控活性。横坐标表示GO的3个模块(生物学过程、 细胞组分、分子功能)。
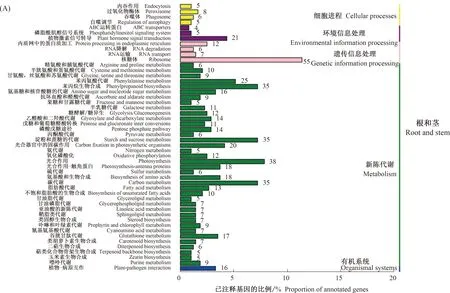
图7 滇黄精器官间差异表达基因的KEGG代谢途径分析图
2.6 滇黄精类黄酮合成基因的筛选
2.6.1 滇黄精类黄酮含量测定结果 滇黄精根、茎和叶中类黄酮含量测定结果如表4所示。滇黄精中不同器官的类黄酮的含量差异较大,从大到小排列的顺序依次是:叶>茎>根,表现出明显的器官特异性。结合根、茎和叶中差异基因的富集情况,发现茎与叶的差异基因数量较少且富集通路分布规律相同,而根与叶和茎的差异基因数量较多,差别较大,推测可能与类黄酮代谢通路中关键酶的合成有关。

表4 滇黄精根、茎、叶中类黄酮的含量
2.6.2 滇黄精类黄酮途径上差异基因的表达 经过数据挖掘,筛选出与类黄酮生物合成相关的10个差异表达基因进行qRT-PCR,发现这些差异基因在器官中均有表达,但规律不尽一致,如图8所示。CHI和CHS在叶中呈现高水平表达,而在根和茎中的表达量较低。DFR、F3’H、C3’H、LAR、CYP73A、CCOMT在根中呈现高水平表达,茎和叶中的表达量较低。ANR在茎中呈现高水平表达,但根和叶中表达量较低。FLS在根、茎和叶中均有表达,器官之间无显著性差异。对滇黄精根、茎和叶中类黄酮的含量与类黄酮途径中10个结构基因进行相关性分析,如表5所示。CHI和CHS与滇黄精器官中类黄酮含量呈显著正相关,说明CHI和CHS是类黄酮生物合成途径中的关键基因,其基因的表达量决定了类黄酮的合成量。
3 结论与讨论
滇黄精是中国著名的药食两用植物,具有健脾补气、降血糖和抗衰老等功效,黄酮类化合物是其主要的活性成分[18]。目前,关于滇黄精的研究主要集中在多糖和甾体皂苷类等途径[19-22],对类黄酮代谢途径的研究却鲜有报道。为了挖掘参与类黄酮生物合成的功能基因,本试验通过高通量测序技术,对滇黄精根、茎和叶进行 RNA-Seq转录组测序并进行组装,最终获得219 769 条unigenes,平均长度为502.31 bp,N50为627 bp,Q30值分别为92.70%、 93.27%和92.54%,均大于90%;获得390 456条转录本,其中在200~1 000 bp范围内的有91.55%,32 976条转录本长度超过2.0 kb。这些结果表明滇黄精转录组序列组装质量较高。将组装后的序列在多个公共数据库中进行注释,共有81 853条unigenes至少在1个数据库中获得功能注释,但仍有137 916条unigenes未被注释,推测与序列片段较短及滇黄精缺少保守的核心序列和基因组信息有关。
本研究通过pathway分析,发现有26 259条(11.9%)unigenes注释到KEGG数据库中,和通路相关的所有基因总数为16 574条。在根和茎中,和通路相关的差异表达基因总数为483条,在根和叶中,和通路相关的差异表达基因总数为560条,在茎和叶中,和通路相关的差异表达基因总数为18条。结合筛选到的 unigenes 在相关数据库中予以的功能注释,进一步确保了所获得基因的可靠性。通过不同器官比较,发现根中的差异基因数量较多,而大多数基因在茎和叶中的表达模式比根中的表达模式更稳定。对差异基因的注释和富集情况进一步分析发现,滇黄精根中高表达基因的功能主要与自身能量储藏代谢相关,茎和叶高表达基因功能主要与叶绿素代谢相关,这一研究结果与器官功能特性相符,说明根的主要功能与物质的吸收和能量储藏有关,茎和叶的主要功能与物质运输、光合作用和呼吸作用等有关。同时,鉴定到与类黄酮生物合成相关的unigenes有46条,以及大量与萜类物质合成相关的unigenes,这也为解析滇黄精黄酮类和萜类物质的合成途径提供了数据支撑。
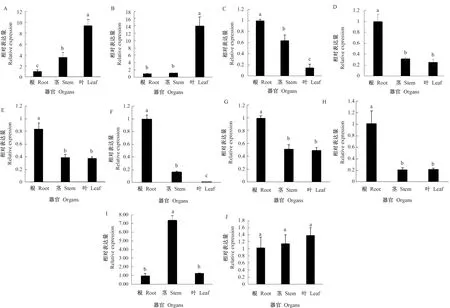
注:A~J为CHS、CHI、DFR、F3’H、C3’H、CCOMT、LAR、CYP73A、ANR和FLS的表达分析。不同小写字母表示差异性显著,显著性水平P<0.05。

表5 滇黄精不同器官中类黄酮含量与类黄酮途径差异表达基因相关性分析
为了进一步解析滇黄精类黄酮合成的分子机制,本研究对滇黄精不同器官中的类黄酮的总含量进行了测定,结果表明不同器官中类黄酮含量存在显著差异,其中叶中类黄酮含量最高,茎次之,根最少,这与北京柠檬[23]、溪黄草[24]、棉花[25]等物种研究结果相一致,表现出明显的器官特异性。基于转录组测序分析,本试验共筛选出10个与滇黄精类黄酮生物合成相关的差异基因,通过qRT-PCR 检测了其在滇黄精不同器官中的表达水平,并与类黄酮含量进行相关性分析,发现CHS和CHI基因表达水平与不同器官中类黄酮含量呈显著正相关关系。在柑橘[23]、银杏[26]等物种中,CHS基因的表达与类黄酮含量变化趋势基本一致。在药用植物盐肤木[27]中,CHI基因的表达与不同器官类黄酮含量也呈良好的正相关关系。这些研究与本研究结果相符,表明CHI和CHS是滇黄精类黄酮生物合成的关键基因,且基因表达水平是导致类黄酮含量差异的重要因素。本研究为滇黄精提供了根、茎和叶的转录组数据,通过对滇黄精类黄酮代谢通路的分析,为挖掘类黄酮生物合成相关基因提供了重要理论依据,也为探究该植物次生代谢途径和更多分子标记奠定基础,有利于将滇黄精作为药用植物资源的开发和利用。
