磁性壳聚糖负载多金属氧酸盐及其催化氧化四氢噻吩的性能
2020-12-09刘叶峰李瑞琪王蕊欣
刘叶峰, 吕 迎, 李瑞琪, 左 鹏, 王蕊欣
(中北大学化学工程与技术学院, 太原 030051)
燃油是目前人类应用最广、用量最大的能源,然而燃油中含硫化合物的燃烧会产生硫氧化物,其中最主要的SOx会导致酸雨、环境酸化、雾霾的形成,此外还会破坏发动机部件、增加颗粒物的排放、加重环境污染、危害人类健康和生存环境[1-3]。目前实现高效、绿色、安全深度脱硫已成为关注焦点。现有的脱硫方法主要有吸附法[4]、萃取法[5]、催化氧化法[6,7]等,其中氧化脱硫因其反应条件温和、高活性、高选择性、低成本等优势是最具前景的脱硫方法之一[8-11]。
多金属氧酸盐(POM)是由早期过渡金属MOx通过共角、共面和共边组装而成的多金属氧配合簇,拥有丰富的拓扑结构和组成,表现出多样的化学性质和物理性质(强酸性、氧化性、光电催化等),并且还具有低温高活性以及无毒、无味、无挥发性等优点,被广泛应用于催化、医学、材料化学等领域[12-15]。近年来,POM独特的催化氧化性能备受关注;但POM作为均相催化剂,存在比表面积小、分离回收困难等缺点,因此选择合适的载体负载POM至关重要。目前常用的载体有大孔树脂、碳纳米管、石墨碳、介孔分子筛(SBA-15)、磁性二氧化硅球等[16-18]。Luo 等[16]制备了功能化氨基改性介孔氧化硅负载型多酸催化剂;Gao等[17]利用浸渍法将POM负载到碳纳米管的通道中,制备的催化剂能使二苯并噻吩的转化率达到99.4%;Ribeiro等[10]利用浸渍法制得了磷钨酸@介孔分子筛复合材料,该催化剂在无乙腈的情况下也具有较高的氧化脱硫性能。
本文以POM对外表面包裹有壳聚糖(CS)的磁性纳米粒子Fe3O4@CS进行表面修饰,制得非均相化催化剂Fe3O4@CS@POM,并将其用于过氧化氢(H2O2)氧化四氢噻吩(THT)的催化过程。所制备的Fe3O4@CS@POM不但具有高催化活性,还同时具有有机修饰磁性Fe3O4微球分散性和稳定性良好的优点,其分离和回收使用也极其便利。
1 实验部分
1.1 试剂和仪器
磷钨酸(H3PW12O40·xH2O,简称 PW12):分析纯,上海麦克林生化科技有限公司;Dawson 型磷钨酸(K6[α-P2W18O62]·14H2O,简称 P2W18)和(K10[α-P2W17O61]·20H2O,简称 P2W17)依据文献 [19, 20]制备;磁性壳聚糖(Fe3O4@CS)微球依据文献[21]制备;THT:分析纯,上海阿拉丁生化科技股份有限公司;其他化学药品和试剂均为市售分析纯。
傅里叶红外光谱(FT-IR)仪:英国,LIantrisant,L1600300 Spectrum Two LITa 型;紫外-可见(UV-Vis)分光光度计:上海尤尼柯公司,2802型;元素分析(EA)仪:青岛华青集团有限公司,PE2400 Series Ⅱ CHNS/O型;扫描透射电子显微镜(TEM):美国,FEI Talos F200S型;激光粒度仪(DLS):德国马尔文公司,Zeta sizer NanoZS型;高效液相色谱(HPLC)仪:北京温分分析仪器技术开发有限公司,LC98-I型;X射线能谱分析(EDS)仪:美国,FEI Super-X型。
1.2 实验步骤
1.2.1 磁性壳聚糖负载化多金属氧酸盐 Fe3O4@CS微球上负载POM:将0.3 g Fe3O4@CS微球加入到30 mL去离子水中,形成A液;将0.6 g PW12(或P2W17、P2W18)溶于15 mL去离子水中,构成B液。将B液缓慢滴加入A液中,室温搅拌12 h后停止反应,以去离子水和无水乙醇依次洗涤,磁分离、干燥得磁性壳聚糖负载化POM 催化剂 Fe3O4@CS@POM(POM=PW12、P2W17或 P2W18)。
1.2.2 Fe3O4@CS@POM催化氧化THT 常压下,以H2O2为氧化剂,将Fe3O4@CS@POM固体催化剂用于THT的催化氧化。将 25.0 μL THT和0.01 g Fe3O4@CS@POM微球分散于5.0 mL乙腈(ACN)中,加入w =30%的H2O2溶液 100 μL ,在25 ℃、100 r/min下恒温振荡105 min,每间隔一段时间取出100 μL溶液溶于4 mL ACN中进行HPLC测试。HPLC的测试条件为:C18反相色谱柱(200 mm×4.6 mm×5 μm),以ACN和去离子水为流动相,0~3 min,/= 20/80;3~12 min,/= 60/40,然后运行 5 min;流速1.0 mL/min;进样量20 μL;柱温为25 ℃;检测波长为210 nm。在此条件下,催化体系中H2O2、环丁亚砜(THTO)和THT的出峰时间分别是3.1、3.4 min和18.4 min。
2 结果与讨论
2.1 非均相催化剂Fe3O4@CS@POM的制备过程
通过微球Fe3O4@CS上外表面包裹的CS上的氨基与POM阴离子之间的静电相互作用,制得表面负载POM的磁性微球Fe3O4@CS@POM。图1所示为Fe3O4@CS@PW12微球的制备过程。

图1 磁性微球Fe3O4@CS@PW12的制备过程Fig. 1 Preparation process of Fe3O4@CS@PW12 magnetic microspheres
2.2 Fe3O4@CS@POM的结构及形貌研究
2.2.1 TEM表征 图2为磁性微球Fe3O4@CS@PW12的TEM照片、EDS以及DLS图谱。由图2(a)可知Fe3O4@CS@PW12微球直径约2 μm,且分散良好。从图2(b,c)中单个小球放大后的图像可看出,微球是由磁性核和厚度为20~30 nm的半透明壳层组成的核壳结构。图2(d)显示Fe3O4的粒径只有326 nm,在包裹了CS并负载磷钨酸形成Fe3O4@CS@PW12后,其粒径明显变大约1 μm,略小于TEM的结果,这可能是由于DLS测试时,粒径较大的球不能稳定悬浮于溶液中所致。图2(e)的EDS能谱图中显示C、O、N、Fe、P、W元素的存在,这说明Fe3O4@CS磁性微球上的确负载了PW12。
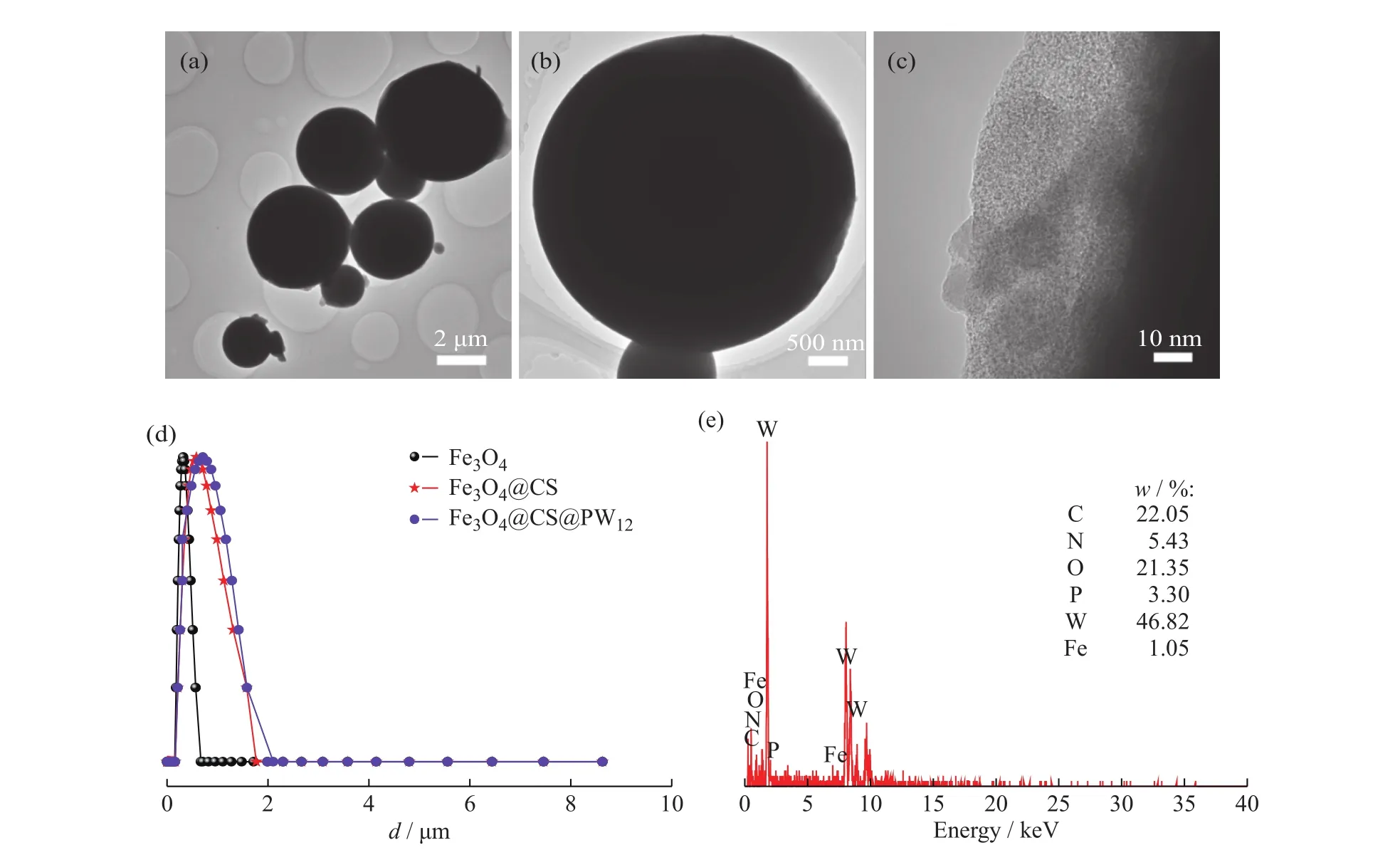
图2 Fe3O4@CS@PW1的(a,b,c)TEM 图,(d)DLS 和(e)EDS 谱图Fig. 2 (a,b,c)TEM images,(d)DLS and(e) EDS spectra of Fe3O4@CS@PW12
2.2.2 光谱分析 图3为磁性微球Fe3O4@CS@PW12的红外光谱图。PW12的谱图中,在1 076~1 088 cm-1、941~979 、880~893 cm-1和 731~782 cm-1处分别出现了 P-Oa、W=Od、W-Ob-W 和 W-Oc-W(Oa是与杂原子配位的四面体氧,Ob和Oc表示共享一个角和一个边的两个八面体的桥接氧原子,Od是每个八面体上的非共用氧)的特征吸收峰。在Fe3O4@CS的谱图中,2 938 cm-1及2 863 cm-1处的峰分别归属于CS上的-CH2和-CH3的伸缩振动和摇摆振动,1 653 cm-1处的峰为CS上的氨基与戊二醛的醛基Shiff反应产生的C=N键的特征吸收峰。而在Fe3O4@CS@PW12的谱图中,除了保留Fe3O4@CS在2 938、2 863 cm-1和1 653 cm-1处的特征吸收峰外,还在700~1 100 cm-1出现了PW12的特征吸收峰,且与纯PW12相比,由于静电相互作用使各吸收峰发生了不同程度的位移,这些吸收峰的出现及变化证明成功制备了Fe3O4@CS@PW12磁性非均相催化剂。
图4为磁性微球Fe3O4@CS@PW12的UV-Vis谱图。在PW12的吸收光谱图上,分别出现了3个吸收峰,在λ=208 nm处出现了端基氧Od→W特征荷移跃迁引起的峰,在λ=273 nm和λ=329 nm处出现了由W-Ob/c(桥氧)-W特征荷移跃迁引起的峰[22],在新制备的磁性微球Fe3O4@CS@PW12的吸收光谱图中,其特征吸收峰与PW12的特征吸收峰基本一致,这说明了PW12成功负载在Fe3O4@CS微球上,且负载后磷钨酸的结构仍然保持不变。

图3 样品的红外光谱图Fig. 3 FT-IR spectra of samples
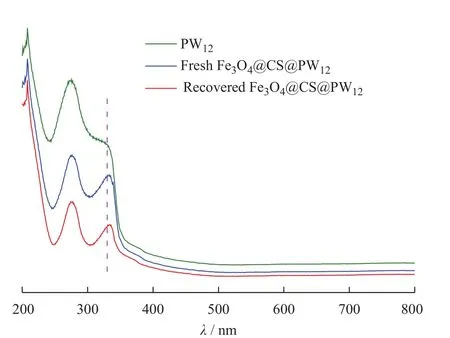
图4 样品的UV-Vis谱图Fig. 4 UV-Vis spectra of samples
2.3 Fe3O4@CS@POM催化氧化THT的性能研究
2.3.1 不同催化体系催化性能的比较 图5(a)示出了常温常压下不同催化体系催化氧化THT的性能,其中催化剂用量为0.01 g、H2O2用量为100 μL。由图5(a)可知,只有Fe3O4@CS@PW12而无H2O2时,THT的转化率为34.56%。
由图5(b)的HPLC图可知,在THT中只加入Fe3O4@CS@PW12后,与初始的THT相比,THT的峰面积变小,然而体系中并未加入THT发生氧化所需的氧源,因此推测THT不能被氧化,且HPLC图中并未出现任何氧化产物峰,这说明溶液中THT的减少并非它本身被催化剂氧化,而可能是被吸附到了Fe3O4@CS@PW12上。
从图5(c)所示的THT氧化产物THTO的产率随时间的变化曲线可知,在THT中加入Fe3O4@CS@PW12后并没有THTO生成,进一步说明溶液中THT的减少是由于Fe3O4@CS@PW12对THT的吸附引起的,并未发生氧化反应,即加入Fe3O4@CS@PW12后THT所显示出的转化率是由Fe3O4@CS@PW12对THT的吸附引起的。
图5(a)显示,在纯H2O2和Fe3O4@CS+H2O2的体系中,THT的转化率分别为28.86%和52.91%,其中Fe3O4@CS+H2O2体系中THT的转化率大于纯H2O2体系的相应值,推测其原因可能是Fe3O4@CS作为载体,其表面的CS对THT有一定的吸附能力,并为H2O2氧化THT提供了良好接触微环境,使氧化过程能够较好地进行。而在同时含有催化剂和双氧水的体系中,THT的转化率明显提高,Fe3O4@CS@PW12+H2O2体系与PW12+H2O2体系相比,前者对THT的催化氧化效果更好,THT的转化率更高,75 min时THT的转化率可高达98.25%,105 min时达100%,且THTO是唯一产物,因此选择性达100%(如图5(d)的HPLC谱图所示)。
根据公式(1)对图5(a)的数据进行准一级动力学方程拟合,结果如图5(e)所示。

式中,k为速率常数,c0为THT的初始浓度,ct为t时刻THT的浓度。
由图5(e)可知,Fe3O4@CS@PW12催化氧化THT符合准一级动力学方程,与文献[23]报道一致,且其动力学常数(0.044 2 min-1)最大,是PW12的动力学常数(0.020 5 min-1)2.16倍,是载体Fe3O4@CS相应值的22.1倍,证明了Fe3O4@CS@PW12非均相催化剂在THT氧化中的催化活性源于PW12簇。

图5 (a)不同体系催化氧化THT的动力学曲线;(b)THT溶液中分别加入Fe3O4@CS@PW12和Fe3O4@CS的HPLC图;(c)THT的氧化产物THTO的产率随时间的变化曲线;(d)Fe3O4@CS@PW12催化THT氧化过程的HPLC图;(e)不同体系催化氧化THT的转化率的拟一级动力学曲线;(f)Fe3O4@CS@PW12及去除催化剂后催化氧化THT的效果图Fig. 5 (a)Kinetic curves of catalytic oxidation of THT in different systems(b)HPLC chromatogram of THT added with Fe3O4@CS@PW12 and Fe3O4@CS, respectively ;(c)Changing curves of THTO yield with time ;(d)HPLC chromatogram of the oxidation process of THT catalyzed by Fe3O4@CS@PW12 ;(e)Pseudo-first-order kinetic curves of the conversion rates of THT catalyzed by different systems ;(f) Oxidation of THT catalyzed by Fe3O4@CS@PW12 and catalyst removal
为了证实非均相催化剂Fe3O4@CS@PW12的非均相本质,我们进行了催化剂分离实验,结果如图5(f)所示。首先,用0.005 g Fe3O4@CS@PW12按照如上的催化过程,按一定时间间隔取样,采用HPLC对反应过程进行检测,当转化率达到57.53%时,将体系中的Fe3O4@CS@PW12催化剂磁分离掉,剩余反应液保持原反应条件继续反应,用HPLC监测反应进程,其转化率随反应时间的变化曲线如图5(f)所示。图中方块图标代表正常的催化反应,当t=165 min 时THT的转化率达84.99%;圆球图标代表催化剂磁分离测试的数据,可以看出,取出催化剂后,40 min 后THT的转化率不再升高,基本保持不变。该磁分离实验证实,Fe3O4@CS@PW12催化剂是作为非均相催化剂催化THT氧化反应的,此外,该催化剂在使用过程中没有Keggin型PW12的脱落,即催化剂结构在催化过程中保持完整。
此外,载体也显示出了一定的环境效应,对脱硫性能的提高起着重要的作用。由于CS在Fe3O4磁性核的外表面包裹,则与CS发生静电相互作用的PW12催化活性位点也暴露于外表面,因此对溶液中的底物具有良好的可接触性,且载体对THT具有良好的吸附性能,这样被吸附到催化剂表面的THT就能够立即与其表面的PW12催化活性中心充分接触,从而很好地发挥催化作用,表现出强催化活性。其催化氧化机理与文献[24, 25]相似,首先催化剂表面的Keggin型磷钨酸与氧化剂H2O2作用,对其进行活化,H2O2对磷钨酸上的WO4八面体构型中的W进行亲核进攻,失去一个水分子,同时磷钨酸被氧化为过氧化多金属氧簇,然后过氧化多金属氧簇将氧原子转移到与之接触的THT上,THT被氧化为THTO,而过氧化多金属氧簇由于失氧又变回多金属氧簇,进行循环反应。Fe3O4@CS@PW12催化氧化THT的机理如图6所示。

图6 Fe3O4@CS@PW12催化H2O2氧化THT的反应机理示意图Fig. 6 Schematic diagram of reaction mechanism of Fe3O4@CS@PW12 catalytic H2O2 oxidation of THT
2.3.2 负载不同类型POM催化剂的催化活性 图7为常温常压下Fe3O4@CS@POM催化氧化THT的效果图。由图可知,3种催化剂中,Fe3O4@CS@PW12的催化氧化效果最佳,反应45 min,THT的转化率达93.69%,105 min达100%。与Fe3O4@CS@PW12相比,Fe3O4@CS@P2W17和Fe3O4@CS@P2W18催化氧化THT的效果较差,反应135 min时THT的转化率分别只有56.12%和58.16%。这与文献[26]规律一致,Keggin型较Dawson型磷钨酸催化氧化脱硫性能好。
2.3.3 影响Fe3O4@CS@PW12催化活性的因素 图8示出了Fe3O4@CS@PW12不同用量下催化氧化THT的动力学曲线。从图中可见,随着催化剂用量增离加,THT的转化率呈先增大后减小的趋势。当Fe3O4@CS@PW12用量由0.005 g增加到0.01 g时,THT的转化率提高较明显,因为较多的催化剂,使催化活性中心数目增多,能够为THT提供更多与催化剂活性中心接触的机会,催化反应速率加快。0.01 g催化剂使THT的转化率达98.25%需75 min,达100%只需105 min,与文献[27]对比,时间大幅缩短;然而,继续增加催化剂用量,相同时间THT的转化率反而下降,这与文献[28]规律一致。因此,该反应体系中Fe3O4@CS@PW12的最佳用量为0.01 g。

图7 THT转化率随反应时间的变化曲线Fig. 7 Curves of THT conversion rate with reaction time

图8 Fe3O4@CS@PW12不同用量时THT的催化氧化曲线Fig. 8 Catalytic oxidation curves of THT with differentFe3O4@CS@PW12 dosages
图9示出了不同H2O2用量下Fe3O4@CS@PW12催化氧化THT的曲线。从图中可以看出,当H2O2用量从25 μL增大到100 μL时,THT转化率也逐渐增大;但当H2O2用量超过100 μL时,随H2O2用量增加,THT转化率反而降低,这是由于H2O2用量增加的同时,H2O的量也相应增加,使催化氧化体系中油水两相的传质阻力变大[29],同时因无效分解造成了H2O2的大量浪费,致使THT的转化率下降。
2.3.4 Fe3O4@CS@PW12的重复使用性 为了进一步证实非均相催化剂Fe3O4@CS@PW12的可重复使用性,每次使用完将催化剂磁分离,用乙醇多次洗涤,干燥后继续投入下次循环使用,其结果如图10所示。从图中可知,当催化剂循环5次后,其THT转化率仍高达96%以上。这说明所制备的催化剂具有良好的稳定性,这对其工业应用至关重要。
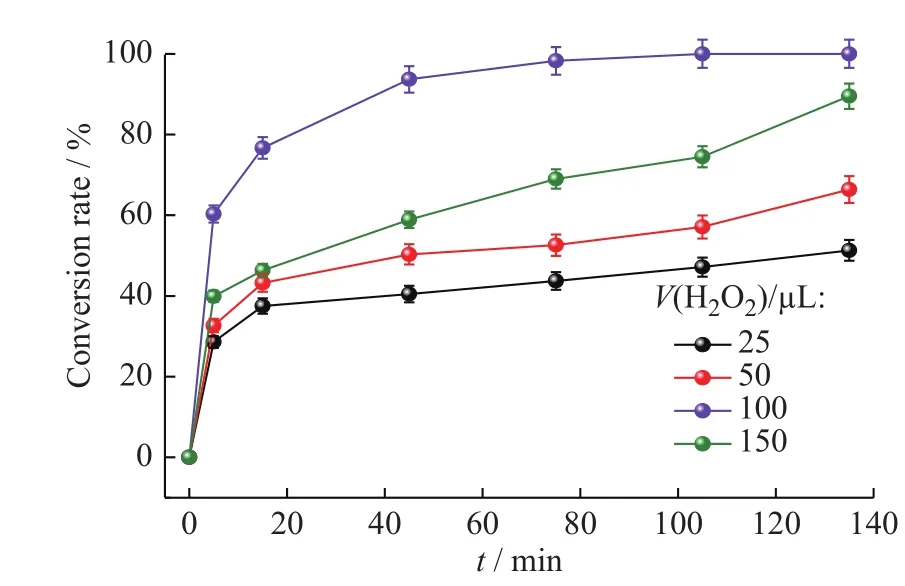
图9 不同H2O2用量时Fe3O4@CS@PW12催化氧化THT的曲线Fig. 9 Catalytic oxidation curves of THT by Fe3O4@CS@PW12 under different H2O2 dosages
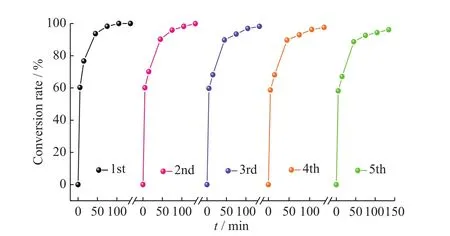
图10 Fe3O4@CS@PW12催化氧化THT的重复使用性Fig. 10 Reusabilities of catalytic oxidation of THT by Fe3O4@CS@PW12
为了考察催化剂的结构稳定性,对循环5次催化氧化结束后的催化剂进行FT-IR和UV-Vis表征(相应的曲线分别示于图3、图4)。回收前后催化剂的红外光谱基本一致,仍保留有PW12在1 076 、972、880 、805 cm-1处的特征吸收峰;回收催化剂的UV-Vis光谱中,PW12在208 、273 nm和329 nm处的吸收峰仍保持不变,这都证明催化剂经多次循环使用后,其结构仍完整。
3 结 论
(1)采用静电结合的方式成功制备了负载POM的磁性微球Fe3O4@CS@PW12、Fe3O4@CS@P2W17和Fe3O4@CS@P2W18。
(2)Fe3O4@CS@PW12的催化效果最好,能够催化 H2O2高效、高选择性地氧化 THT为 THTO。Fe3O4@CS@PW12和H2O2的用量对THT的催化氧化性能有较大影响,两者的用量过大或过小都不利于催化氧化性能的发挥。常温常压下,0.01 g Fe3O4@CS@PW12(PW12的负载量为57.2%)、100 μL H2O2条件下,105 min便能使25 μL THT的转化率达100%,氧化产物THTO的选择性也达100%。
(3)Fe3O4@CS@PW12经5次重复使用后,催化活性没有明显降低,且催化剂结构保持完整。
(4)催化剂的分离、回收操作简单易行,通过简单的磁分离操作即可回收使用,为其工业应用奠定了坚实基础。
