裸花紫珠药材及相关制剂质量控制方法研究
2020-11-18付辉政任琦周志强陈伟康魏峰马双成罗跃华
付辉政 ,任琦,周志强,陈伟康,魏峰,马双成,罗跃华*
1.江西省药品检验检测研究院,国家药品监督管理局中成药质量评价重点实验室,江西省药品与医疗器械质量工程技术研究中心,南昌 330029;2.中国食品药品检定研究院,北京 100050
裸花紫珠(Callicarpa nudifloraHook.ex Arn.)为马鞭草科(Verbenaceae)紫珠属植物,主产于广东、广西、海南等地[1],为海南大宗地道药材,目前在江西赣州、吉安、抚州等地进行了广泛的栽培,具有散瘀消肿、抗菌止血、消炎解毒、驱风祛湿之功效。临床上用于治疗化脓性炎症、急性传染性肝炎、呼吸道及消化道出血、创伤出血等症,外用治烧、烫伤及外伤出血等[2],被誉为天然消炎止血圣药。研究表明裸花紫珠主要含有黄酮类、萜类、苯丙素类和挥发油等成分,具有止血、抗血栓、抗炎、抑菌、细胞毒活性、增强免疫等多种药理活性[3-8],但对于裸花紫珠基础研究还很薄弱,尤其是其药效物质及作用机理还尚不明确。
裸花紫珠最早收载于《中国药典》1977 年版[9],标准项下仅有性状、理化鉴别和检查项,标准较低。最早由裸花紫珠单味药材制成的裸花紫珠片,收载于卫生部药品标准《中药成方制剂》第六册,标准项下仅有性状、理化鉴别、检查质控项目,难以有效控制产品的质量。裸花紫珠胶囊和颗粒系由裸花紫珠片通过工艺革新创制而成,其原标准中质控项目简单且缺乏专属性。为了更好地控制裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒质量,笔者针对裸花紫珠药效物质基础和作用机制尚不明确问题,对裸花紫珠化学成分及药理活性进行了系统研究,从裸花紫珠醇提物中分离鉴定了76个化学成分,其中新化合物21 个,通过体内、外药理研究,系统阐明了裸花紫珠药效物质基础及作用机理,并明确了可用于裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒质控定量活性指标成分木犀草苷和毛蕊花糖苷。在此基础上,对裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒的鉴别、指纹图谱、含量测定质量控制方法以及主要活性成分药代动力学进行了研究,建立了裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒5 项国家药品标准,形成了从裸花紫珠-中间体-系列制剂的完整质量控制体系,保障了产品质量,同时也为指导临床合理用药提供了科学依据。
1 材料
岛津2010 系列高效液相色谱仪、Aglient Technologies 1260 高效液相色谱仪、岛津2010 型制备高效液相,检测器均为DAD 检测器;Perkin-Elmer Lambda 25 MV-Vis 分光度计;Perkin-Elmer 341 旋光仪;MDS SCIEX APCI 2000 LC-MS-MS 测定ESI-MS 由;Waters ACQMITY MPLC/Xevo G2 Q TOF测定HRESIMS,Bruker AVANCE III 600 超导核磁共振仪测定1H-NMR (600MHz)和13C-NMR (150MHz)谱及二维谱(HSQC,HMBC,1H-1H COSY,NOESY)谱。柱层析硅胶、薄层层析硅胶(青岛海洋化工厂);乙腈、甲醇为色谱纯;水为 Milli-Q 制备的超纯水、其他所用试剂均为分析纯。SD 大鼠,体重(200±20) g,购自湖南斯莱克景达实验动物有限公司(许可证号SCXK[湘]2009-0004),动物房温度(22±2)℃,湿度55%±10%,每日光照时间12 h;动物饲料购自北京科澳协力饲料有限公司。对照品咖啡酸(批号110885-200102)、木犀草素(批号111520-200201)、毛蕊花糖苷(批号1530-200202)、木犀草苷(批号11720-201106)、连翘酯苷B(批号111811-201102)均购于中国食品药品检定研究院。其余对照品均由江西省药品检验检测研究院从裸花紫珠中分离制备得到,由MS、1H-NMR和13C-NMR 鉴定结构,通过面积归一化法计算纯度均在98.0 %以上。裸花紫珠采自海南省五指山区,经江西省药品检验检测研究院罗跃华主任中药师鉴定为马鞭草科紫珠属裸花紫珠C.nudifloraHook.的叶。裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒药品的来源信息见表1。
2 方法与结果
2.1 裸花紫珠物质基础研究
利用硅胶、Sephadex LH-20、MPLC、分析和制备HPLC 等分离分析技术和方法对裸花紫珠化学成分进行了分离纯化[10-14],根据化合物的理化性质、质谱、1D NMR 和2D NMR 等波谱分析技术鉴定了76 个化合物,发现新化合物21 个(见图1),46个化合物为首次从裸花紫珠中分离得到,系统地阐明了裸花紫珠的化学成分,为裸花紫珠质量控制研究奠定了基础。


图1 裸花紫珠中21个新化合物结构
2.2 裸花紫珠药效物质作用机理研究
建立了反映裸花紫珠止血、活血、抗炎等5种动物模型和68 种指标的现代药理学方法,对裸花紫珠提取物及分离得到的单体化合物进行系统药理药效评价,结果表明:1,6-di-O-caffeoyl-β-D-glucopyranoside 和木犀草素-3'-O-β-D-吡喃葡萄糖苷可降低花生四烯酸诱导的血小板聚集率和血小板中P-选择素、TXA2的含量,显示较强抗花生四烯酸诱导血小板聚集活性,IC50值分别为0.129 mmol/L 和0.041 3 mmol/L(阳性药奥扎格雷终浓度为0.2 mmol/L 时,抑制率为43.80%)。其体外机理是通过抑制血小板PI3K/Akt 信号通路,1,6-di-O-caffeoyl-β-D-glucopyranos-ide 同时具有抑制PLC/PKC 信号通路的作用,从而抑制蛋白RhoA、PKA 的表达,降低血小板聚集率、TXA2的生成和P-选择素的释放,贯穿血小板形变、聚集、释放等多个反应;2α,3α-二羟基-12-烯-28-乌苏酸和(3S,5R,6R,7E,9S)-megastigmane-7-ene-3,5,6,9-tetrol 对MDA-MB-231 和MDAMB-468 细胞的体外侵袭和迁移能力具有明显地抑制作用,并对高转移乳腺癌细胞的体外迁移、侵袭和粘附能力均有显著抑制作用,其作用机理是通过抑制细胞骨架蛋白F-actin 的聚合而下调肿瘤细胞的体外运动能力,并可降低PI3K/Akt 通路上PI3K110β 等6 个蛋白的表达,提示上述两个化合物可能是通过下调 PI3K/Akt 通路信号转导,干扰细胞骨架蛋白F-actin 的形成,从而发挥抑制肿瘤细胞的体外运动能力;木犀草素、木犀草苷、毛蕊花糖苷显示较强的抗氧化活性;木犀草苷、木犀草素-3'-O-β-D-吡喃葡萄糖苷、木犀草素-4'-O-β-D-吡喃葡萄糖苷、木犀草素、5,4'-二羟基-3,7,3'-三甲氧基黄酮、5-羟基-3,7,3',4'-四甲氧基黄酮、毛蕊花糖苷均显示出不同程度的抑制LPS 诱导RAW264.7 细胞NO 生成作用。上述研究系统地验证了裸花紫珠的功效,并深入阐明了裸花紫珠药效物质及作用机理,也为裸花紫珠、干浸膏、制剂质控指标选择提供了依据。
2.3 裸花紫珠药材、干浸膏、裸花紫珠片胶囊颗粒共有成分方法研究
采用UPLC-ESI-Q-TOF-MSE分析方法(见图2),根据质谱数据以及对照物质比对,鉴定了裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒中共有的13个指标成分结构,依次分别为:柯伊利素-7-O-β-D-吡喃葡萄糖苷、nudifloside、木犀草苷、连翘酯苷B、毛蕊花糖苷、异毛蕊花糖苷、木犀草素-4'-O-β-D-吡喃葡萄糖苷、木犀草素-3'-O-β-D-吡喃葡萄糖苷、7-O-E-p-coumaroyl-gardoside、木犀草素、5,4'-二羟基 -3,7,3'-三甲氧基黄酮、2α,3β,19α-三羟基-12-烯-28-乌苏酸、异鼠李素。具体的方法条件如下,色谱柱:Waters ACQUITY UPLC®BEH-C18(2.1 mm×100 mm,1.7 μm);流动相:甲醇(A)-甲酸铵甲酸水溶液(每1 000 mL 水中含1 mL 甲酸和2 mmol 甲酸铵)(B);梯度洗脱:0~17 min,5%→70% A;17~27 min,70%→80% A;流速:0.4 mL/min;柱温:60 ℃;进样量:2 μL。采用电喷雾电离离子源(ESI),Leucine-enkephalin 作校准液,负离子模式检测,m/z 100~1 000,采样间隔为0.5 s,毛细管电压为2.5 kV,锥孔电压为40 V,离子源温度为100 ℃,脱溶剂温度为450 ℃,脱溶剂气体流速800 L/h,锥孔气流量 40 L/h,碰撞气体为氩气;碰撞低能量为6 V,碰撞高能量20 V~40 V。初步阐明了裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒的药效物质组成,为裸花紫珠制剂的质量控制提供了依据,同时确定了主要量大活性成分木犀草苷和毛蕊花糖苷为裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒定量质控指标。
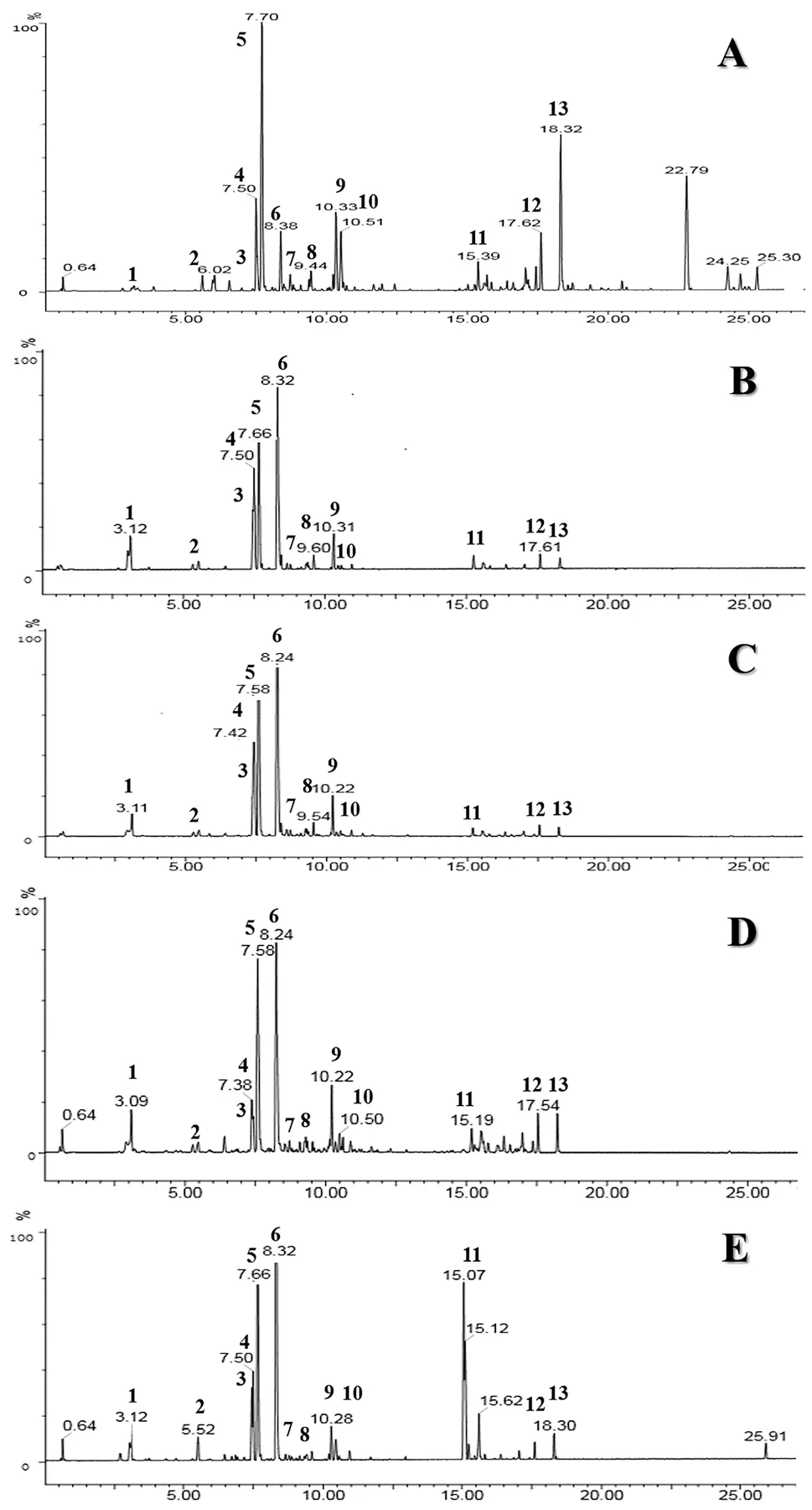
图2 负离子模式下各样品UPLC-ESI-Q-TOF-MSE BPI图
2.4 裸花紫珠药材、干浸膏、裸花紫珠片胶囊颗粒定性方法研究
2.4.1 显微鉴别采用显微法建立了裸花紫珠叶横切面和表面观显微鉴别,具体如下:叶横切面:上下表皮均为1 列细胞,外均被腺毛、非腺毛和腺鳞,以下表皮较多;上表皮下方有一列较大的下皮细胞。栅栏组织为1~2 列细胞;海绵组织细胞小,排列较紧密。主脉维管束外韧型,呈马蹄状环,韧皮部有纤维群。主脉上下表去皮内侧均有数列厚角细胞。薄壁细胞草酸钙簇晶或方晶。见图3。
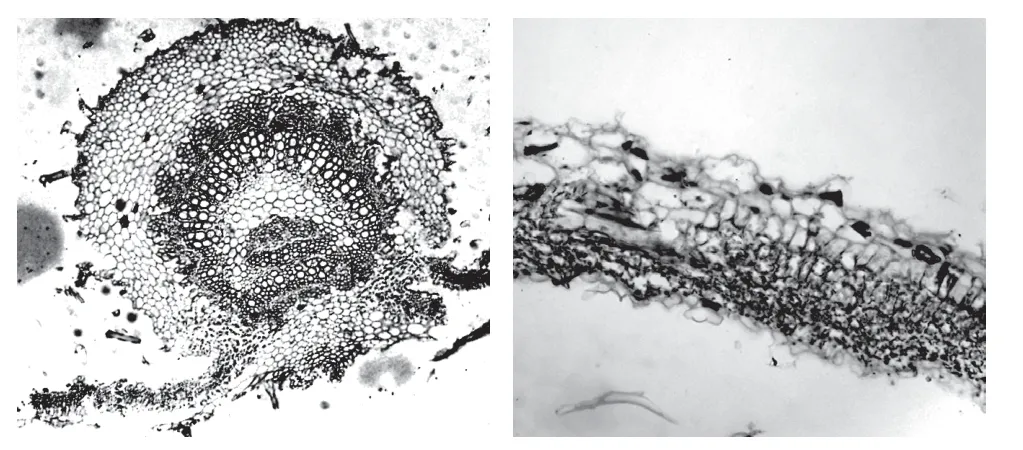
图3 裸花紫珠横切面永久制片图
叶表面观:非腺毛有两种:一种为迭生星状毛,大多碎断,中轴直径18~30μm,壁厚,非木化,完整者1~10 余轮;每轮侧生1~7 个侧生细胞。另一种1~4 个细胞,末端有分叉,壁薄。腺鳞头部6~8 个细胞,扁球形,直径50~60 μm。腺毛头部4个细胞,直径22~27 μm,柄1~2个细胞。上皮细胞多角形,壁略呈连珠状增厚。气孔不定式,保卫细胞长约25 μm。见图4。

图4 裸花紫珠叶表面观显微图
2.4.2 薄层色谱鉴别采用薄层色谱法分别建立了裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒薄层色谱鉴别,具体如下:供试品溶液制备:取裸花紫珠1.0 g;或取裸花紫珠干浸膏0.7 g;或取裸花紫珠片,除去包衣,研细,取0.8 g;或取裸花紫珠胶囊内容物1.0 g;或取裸花紫珠颗粒2.5 g,研细。加水150 mL,煎煮,保持微沸1 h,放冷,滤过,滤液加氯化钠5 g,振摇使溶解,溶液加乙酸乙酯40 mL 振摇提取,分取乙酸乙酯液,蒸干,残渣加甲醇1 mL 使溶解,作为供试品溶液;对照药材溶液制备:取裸花紫珠对照药材1 g,按供试品溶液的制备方法制成对照药材溶液;色谱条件及系统适用性试验:采用0.5 %氢氧化钠溶液制备的硅胶G 薄层板;点样量:10 μL;展开剂:乙酸乙酯-甲醇-浓氨水(17∶2∶1);显色剂:喷以3%三氯化铝乙醇溶液,在105 ℃加热5 min,置紫外光灯(365 nm)下检视。结果:供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。通过多次试验,重复性好,阴性样品无干扰。见图5。

图5 各样品薄层色谱图
2.5 裸花紫珠药材、干浸膏、裸花紫珠片胶囊颗粒HPLC定量方法研究
采用HPLC 多波长切换技术建立了同时测定裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒中木犀草苷和毛蕊花糖苷的含量测定方法,具体如下:供试品溶液制备:取裸花紫珠粉末(过四号筛)约1.0 g;或取裸花紫珠干浸膏,研细,取约0.7 g;或取装量差异项下裸花紫珠片,除去薄膜衣,精密称定,研细,取约0.8 g;或取装量差异项下裸花紫珠胶囊内容物,研细,取约1.0 g;或取装量差异项下裸花紫珠颗粒,研细,取约2.5 g。精密称定,置具塞锥形瓶中,精密加入70 %甲醇50 mL,称定重量,超声处理(功率500 W,频率40 kHz)40 min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,作为木犀草苷测定用供试品溶液。另精密量取续滤液5 mL,置50 mL 量瓶中,加70%甲醇稀释至刻度,摇匀,作为毛蕊花糖苷测定用供试品溶液;对照品溶液制备:取木犀草苷对照品、毛蕊花糖苷对照品适量,精密称定,分别加70%甲醇制成每1 mL 含木犀草苷20 μg、毛蕊花糖苷40 μg 的溶液,即得;色谱条件及系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相:乙腈(A)-0.1%甲酸(B),梯度洗脱:0~50 min,14% A;50~51 min,14% →80% A;51~61 min,80% A;流速:1.0 mL/min;进样量:10 µL;柱温为35℃;木犀草苷检测波长为350 nm,毛蕊花糖苷检测波长为330 nm。结果:木犀草苷的含量在2.63~131.30µg/mL 呈良好的线性关系,Y木犀草苷=26.87X-7.204(r=1.000 0);毛蕊花糖苷的含量在3.88~194.2 µg/mL呈良好的线性关系,Y毛蕊花糖苷=16.09X-4.979(r=0.999 7)。木犀草苷加样回收率为100.1%,RSD 为1.4%(n=6);毛蕊花糖苷加样回收率为100.4%,RSD 为1.4%(n=6),样品结果见表1 和图6。本方法简单、准确,灵敏度高,重复性好,可用于裸花紫珠、干浸膏、裸花紫珠片/胶囊/颗粒的质量评价。

图6 木犀草苷对照品、供试品及阴性样品(A)和毛蕊花糖苷对照品、供试品及阴性样品(B)HPLC色谱图

表1 样品含量测定结果
2.6 裸花紫珠药材、干浸膏、裸花紫珠片胶囊颗粒HPLC指纹图谱方法研究[15]
建立了裸花紫珠药材、干浸膏、片剂、胶囊、颗粒的HPLC 指纹图谱方法。具体如下:供试品溶液制备:取样品粉末(过4 号筛)约1.0 g,精密称定,置100 mL 具塞锥形瓶中,精密加入70%甲醇50 mL,称定重量,超声处理(功率500 W,频率 40 kHz)40 min,放冷,再次称定重量,并用70%甲醇补重,摇匀,滤过,取续滤液,即得;对照品溶液制备:精密称取咖啡酸、连翘酯苷B、nudifloside、木犀草苷、毛蕊花糖苷、异毛蕊花糖苷、juncein、7-O-E-p-coumaroyl-8-epi-lo-ganic acid、木犀草素、5,4'-二羟基-3,7,3'-三甲氧基黄酮对照品适量,置10 mL 量瓶中,加70%甲醇溶解并稀释至刻度,摇匀,即得;色谱条件及系统适用性试验:采用Waters Sunfire C18 色谱柱(250 mm×4.6 mm,5 µm);流动相:乙腈(A)-0.1%甲酸(B);梯度洗脱:0~50 min,10%→20% A;50~70 min,20%→40% A;70~90 min,40%→80% A;流速:1.0 mL/min;检测波长:330 nm;柱温:30 ℃;进样量:10 µL。因毛蕊花糖苷色谱峰峰面积大且峰形较好,故以其为参照峰,计算各批次样品共有峰的相对保留时间,显示其RSD 均符合指纹图谱的要求。取10 批海南五指山代表性药材色谱图导入中药色谱指纹图谱相似度评价系统(2012 版),建立对照指纹图谱,指认了13 个共有峰(见图7)。将其他批次样品的色谱峰与参照图谱进行自动匹配,结果:各批次裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒的指纹图谱相似度较高,相似度均在0.9 以上。见图8。

图7 裸花紫珠13个共有峰

图8 裸花紫珠(A)、裸花紫珠干浸膏(B)、裸花紫珠片(C)、裸花紫珠胶囊(D)、裸花紫珠颗粒(E)质量一致性对比图
2.7 UHPLC-MS/M法同时测定大鼠血浆中裸花紫珠7个活性成分及其药动学研究[16]
建立UHPLC-MS/MS 法同时测定大鼠血浆中裸花紫珠提取物7 个活性成分木犀草苷、dracocephaloside、Juncein、毛蕊花糖苷、异毛蕊花糖苷、连翘酯苷B、nudifloside 的血药浓度,并研究提取物单次给药后大鼠体内的药代动力学过程。方法:大鼠灌胃给予裸花紫珠提取物,分别于给药后5、10、20、30、45、60、90、120、180、300、420 min 经眼内眦静脉丛取血0.4 mL 置于肝素化离心试管中,依次加入10 μL 内标,50 μL 盐酸(0.25 mol/L),10 μL 甲醇,涡旋混匀,加入乙腈300 μL,涡旋灭活蛋白。离心,取上清液于40 ℃水浴下氮气吹干,残渣加50%乙腈100 μL 复溶,涡旋混匀后离心,取上清液,采用内标法(SMZ),色谱柱:Phenomenex® Kinetex-C18(50 mm×2.1 mm,1.7 μm);流动相:乙腈(A)-0.5%甲酸(B)梯度洗脱:0~7.0 min,10%→43% A;7.0~7.1 min,43%→95% A;7.1~9.0 min,95% A;柱温:40 ℃;流速:0.45 mL/min。质谱条件:电喷雾离子源(ESI);负离子方式检测;MRM(多重反应监测)模式;气帘气(N2):25 psi;离子源GS 1(N2):60 psi,GS 2(N2):55 psi;源内温度500 ℃;喷雾电压:-4 500 V;入口电压(EP):-10 V;出口电压(CXP):-13 V;7 种活性成分的母离子、子离子m/z 分别为(447.1/285.0)、(447.1/285.0)、(447.1/285.0)、(447.1/285.0)、(623.3/161.0)、(623.3/161.0)、(755.2/160.7)、(523.4/160.9)、(417.2/254.7)。结果:空白生物样品的内源性物质对7 种活性成分和内标物测定无干扰(见图9)。

图9 空白血浆(A)、空白血浆+对照品(B)和给药20 min后的血浆样品(C)MRM色谱图
血浆中7 种活性成分的线性范围分别为2.06~1 030 ng/mL、2.32~1 160 ng/mL、1.65~825 ng/mL、7.77~3 880 ng/mL、5.04~2 520 ng/mL、1.78~890 ng/mL、1.75~875 ng/mL,定量下限分别为1.03 ng/mL、1.16 ng/mL、0.82 ng/mL、3.88 ng/mL、2.52 ng/mL、0.89 ng/mL、0.88 ng/mL,线性关系好(r均大于0.995)。日内、日间精密度(RSD)均小于8.8%,准确度(RE)在-9.6%~9.8%之间。方法的绝对提取回收率均大于75%;基质效应低;稳定性研究结果表明样品稳定;7 种活性成分药学动力学参数如下:T1/2分别为25.39±1.05、26.04±2.02、26.49±0.74、235.41±117.90、151.56±49.23、161.68±63.92、42.58±39.55 min;AUC0-t分别为3 134.65±413.45、2 944.69±571.63、2 801.90±304.44、93 881.65±18 326.65、29 204.97±8 499.88、15 027.05±3 763.82、987.50±232.30 ng/mL·min;AUC0-∞分别为3 134.68±413.46、2 944.72±571.64、2 801.94±304.44、100 776.25±25 227.72、30 006.63±8 709.22、16 237.88±3 872.06、1 002.13±266.55 ng/mL·min;Cmax分别为67.55±8.67、59.55±7.24、52.10±4.07、2 179.00±355.60、737.57±210.31、227.30±48.38、17.16±3.36 ng/mL,7 种活性成分均在30 min 左右达到最大血药浓度,后又被快速地消除(见图10)。

图10 大鼠ig裸花紫珠提取物后7种活性成分的药时曲线
3 讨论
本课题组对裸花紫珠物质基础及作用机理进行系统研究。从裸花紫珠中共分离鉴定了76个化合物,其中新化合物21 个,46 个化合物为首次从裸花紫珠中分离得到。建立了反映裸花紫珠止血、活血、抗炎等5 种动物模型和68 种指标的现代药理学方法,并明确了可用于裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒质控定量活性指标成分木犀草苷和毛蕊花糖苷。
建立了UHPLC-MS/MS 同时测定大鼠血浆中裸花紫珠提取物7 种活性成分的血药浓度的方法,从药-时曲线图中可看出7 种活性成分均在30 min 左右达到最大血药浓度,后又被快速地消除,这可能与其相对较大的极性有关。木犀草苷、dracocephaloside、juncein 表现出一致的药动学行为;毛蕊花糖苷、异毛蕊花糖苷也表现出类似的药动学行为;木犀草苷与毛蕊花糖苷的药-时曲线与文献报道基本一致[17];而裸花紫珠提取物中连翘酯苷B 的药动学规律与在其他提取物中略有不同[18],其原因可能是裸花紫珠中众多化合物的相互作用的结果所造成的。虽然nudifloside 的给药剂量高于黄酮苷类化合物,但其AUC0-t低于黄酮苷类,这表明nudifloside 的生物利用度低于黄酮苷类。
建立了裸花紫珠药材显微鉴别及裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒的专属性强的TLC 鉴别方法;建立了裸花紫珠药材、干浸膏、的HPLC 指纹图谱方法;采用HPLC 多波长切换技术建立了同时测定裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒中木犀草苷和毛蕊花糖苷的含量测定方法。最终起草了裸花紫珠药材、干浸膏、裸花紫珠片/胶囊/颗粒5 项国家药品标准,形成了从裸花紫珠-中间体-系列制剂的完整质量控制体系,为临床合理用药提供可参考的科学依据。
