蜂毒肽在大肠杆菌中的高效融合表达与纯化
2020-11-11周丽仙张荣珍李利宏
周丽仙 , 张荣珍 *, 李利宏 , 徐 岩
(1. 江南大学 生物工程学院,江苏 无锡 214122;2. 江南大学 工业微生物教育部重点实验室,江苏 无锡 214122)
蜂毒肽来自于欧洲蜜蜂(Apis mellifra),是蜜蜂毒液中的主要毒性成分,是一种重要的阳离子生物活性肽,具有很好的抗菌[1-3]、抗炎症[4-7]、抗肿瘤[8-11]和抗艾滋[12]等生物功能。
蜂毒肽由 26 个氨基酸残基组成 (NH2-GIGAVLKVLTTGLPALISWIKRKRQQ -CONH2), 其中疏水性主要集中在氨基末端区域(残基1~20),而亲水性主要集中在含有一段带正电荷的氨基酸残基的羧基末端区域(残基21~26)[13]。 蜂毒肽因具有两亲性质而具有水溶性,容易结合在带负电荷的膜表面[14],对膜具有强扰动活性,通过形成跨膜孔或离子通道或以表面活性剂的活性方式作用,伴随着原子、离子和分子的泄漏以及膜渗透性的增强来干扰磷脂双分子层的完整性[15-17]。 在蜜蜂的生物合成过程中,首先形成含信号肽和前导肽的前体蛋白——前蜂毒肽(proMET),经过剪切前导肽和信号肽后释放形成蜂毒肽(MET)[18]。 由于蜂毒肽前体蛋白富含酸性氨基酸和脯氨酸,且前结构域带有高度负电荷而赋予分子整体负电荷,因而比蜂毒肽具有更大的溶解度,也因此降低了对细胞膜的破坏能力[10]。
制备蜂毒肽主要有3 种途径:从天然蜜蜂毒液中直接分离纯化[19]、化学合成法以及生物法制备。目前, 蜂毒肽的获得方式主要是从蜂毒中分离提纯,或者通过化学合成的方式来获得。 由于蜂毒肽只占蜂毒干质量的50%,从蜂毒中分离大量蜂毒肽存在极大的局限性。 且分离纯化蜂毒肽首先要获得蜂毒,生产中常用电刺激蜜蜂采取蜂毒,往往以蜜蜂生命为代价,不利于蜜蜂群势发展和养蜂生产。 并且由于蜂毒成分复杂,存在多种蛋白质,且各组分相对分子质量接近,尤其是与蜂毒肽相对分子质量接近的磷脂酶A2,造成了纯化上的困难、操作的繁琐以及高成本, 使蜂毒肽的广泛应用受到了限制。化学合成法主要是以氨基酸为原料,通过固相合成法等方法合成多肽或蛋白质,具有污染严重且目标蛋白质得率低等缺点。
生物法制备蜂毒肽,由于蜂毒肽相对分子质量只有2 900 且不容易活性表达, 同时在蜂毒肽的纯化过程中也存在困难。 因为蜂毒肽表现出对细胞高效的致死性, 并且容易被细菌体内的蛋白酶降解,因而直接在原核系统中难以表达。 因此充分开发利用生物法制备蜂毒肽,具有很好的社会经济效益和应用前景。
作者选用麦芽糖结合蛋白基因与蜂毒肽基因进行融合表达,获得高效可溶性表达的蜂毒肽融合蛋白。 融合表达具有多方面的优点:实现外源基因的高效转译, 有助于减少或防止包涵体的产生,促进蛋白质的正确折叠,限制蛋白酶降解和有助于纯化等。 麦芽糖结合蛋白 (maletose-binding protein,MBP)是大肠杆菌中麦芽糖积累途径里一个重要的蛋白质,有研究报道 MBP 具有强助溶作用,可以显著提高与之融合的外源蛋白质的可溶性表达[20]。 将麦芽糖结合蛋白质基因分别克隆在前蜂毒肽基因、蜂毒肽基因N 端,并插入6 个组氨酸标签,构建在质粒pET15-b 上,在大肠杆菌中能够高效可溶性表达目标蛋白质。 经两步亲和纯化,得到了纯度高达90%以上的融合蛋白质, 通过牛津杯抑菌实验确定了融合蛋白质的抑菌功能。 该制备方法为生物法制备蜂毒肽奠定了较好的研究基础。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 质粒pET15b、MBP3、大肠杆菌(E. coli)JM109 和 BL21(DE3)菌株:江南大学酿造微生物学与应用酶学研究室保存;promet 基因:由生工生物工程(上海)股份有限公司(Sangon)合成;质粒pMD19-T: 购自宝生物工程 (大连) 有限公司(Takara);引物合成和测序:由金唯智生物科技有限公司(苏州)完成。
1.1.2 主要试剂 氨苄青霉素、 异丙基-β-D-硫代半乳糖苷 IPTG: 购自 Sangon 公司;PrimeStar®Max(Premix)、rTaq DNA polymerase for PCR、限制性内切酶 BamH I 和 Xho I、T4 DNA 连接酶、DNA marker、蛋白质相对分子质量marker: 购自 Takara 公司;PCR 产物纯化试剂盒、DNA 胶回收试剂盒、 质粒抽提试剂盒:购自Omega 公司。
1.1.3 主要仪器 Ni 柱 (5 mL)、Dextrin Sepharose HP 亲和吸附柱 (1 mL),ÅKTA purifier 蛋白质纯化系统:购自美国GE Healthcare 公司。
1.2 方法
1.2.1 PCR 引物的设计与合成根据Dmitry S.Perekalin等[21]报道的蜂毒肽氨基酸序列(含信号肽和前导肽)进行密码子优化化学合成promet 基因, 并设计引物,序列见表 1。 上游引物 PF1、PF2,引入 His6 基因, 下游引物PR2 引入Xho I 酶切位点(用下划线表示)。 以质粒MBP3 为模板设计引物, 上游引物PF3,5‘引入 Nco I 酶切位点,下游引物 PR2。
1.2.2 met、promet 基因与 mbp 基因的扩增 以化学合成的promet 基因为模板, 由引物对PF1/PR1、PF2/PR1 经 PCR1 分别扩增 MET 片段和 proMET 片段。 PCR 条件设为:98 ℃预变性 30 s,以 98 ℃变性15 s,55 ℃退火 15 s,72 ℃延伸 20 s, 进行 30 个循环,72 ℃保温10 min;以质粒MBP3 为模板,由引物PF3、PR2 进行 PCR2 扩增 MBP 片段。 PCR 条件设为:98 ℃预变性 30 s, 以 98 ℃变性 15 s,55 ℃退火15 s,72 ℃延伸 1 min 10 s,进行 30 个循环,72 ℃保温10 min; 以 MET 片段和 MBP 片段为模板,由引物对PF3/PR2 重叠延伸扩增MBP-MET 片段,以proMET 片段和MBP 片段为模板, 由引物对PF3/PR2 重叠延伸扩增 MBP-proMET 片段。PCR 条件设为:98 ℃预变性 30 s, 以 98 ℃变性 15 s,55 ℃退火15 s,72 ℃延伸 1 min 20 s,进行 10 个循环。 加入引物 PF3 和 PR2 再进行 20 个循环,72 ℃保温10 min。PCR 产物用1 g/dL 的琼脂糖凝胶电泳检测。PCR 产物纯化后用T4 DNA 连接酶与载体pMD19-T 于16 ℃连接4 h, 连接产物转化E. coli JM109 感受态细胞,100 μg/mL 氨苄青霉素(Amp)抗性 LB 平板筛选阳性克隆,提取的质粒经琼脂糖凝胶电泳及测序鉴定, 质粒分别命名为 pMD19T-MBP-MET 和pMD19T-MBP-proMET。

表1 引物Table 1 Primers
1.2.3 重组表达载体pET-MBP-MET 和pETMBP-proMET 构建 限制性内切酶Xho I 和Nco I 分别双酶切重组克隆质粒pMD19T-MBP-MET 和pMD19T-MBP-proMET, 琼脂糖凝胶回收插入片段MBP-MET 和MBP-proMET, 分别与用相同限制酶双酶切的表达载体pET15-b 过夜连接,连接产物转化E. coli JM109 感受态细胞,涂布 100 μg/mL Amp抗性LB 平板,挑取单菌落抽提质粒进行测序,序列正确的质粒分别命名为pET-MBP-MET 和pETMBP-proMET,构建示意图见图1。

图 1 标签融合的 pET-MBP-MET、pET-MBP-proMET质粒构建示意图Fig. 1 Schematic representations of the tag-fused pETMBP-MET, pET-MBP-proMET constructs
1.2.4 融合蛋白质的诱导表达 分别转化重组表达 质 粒 pET-MBP-MET、pET-MBP-proMET 至E. coli BL21(DE3)感受态,涂布于含有 100 μg/mL Amp 的 LB 的固体培养基上,37 ℃培养过夜, 挑取单菌落接种于含有100 μg/mL Amp 的LB 的液体培养基中,37 ℃、200 r/min 振荡培养 10 h 后, 按体积分数1%的转接至100 mL 新鲜的含有100 μg/mL Amp 的 LB 的液体培养基中,37 ℃、200 r/min 振荡培养至OD600约为0.6 时,添加终浓度为0.1 mmol/L的诱导物异丙基-D-硫代半乳糖苷(IPTG),于30 ℃、200 r/min 诱导培养16 h。 4 ℃下离心收集菌体细胞,超声破碎后收集离心的上清液及沉淀,加入4×SDS-PAGE loading buffer 煮沸 10 min, 用 9%SDSPAGE 进行验证。
1.2.5 融合蛋白质MBP-MET 和MBP-proMET 的纯化 重组大肠杆菌E. coli BL21/pET-MBP-MET、E. coli BL21/pET-MBP-proMET 分别在上述条件诱导培养后,将500 mL 发酵菌液离心收集菌体,加入50 mL Ni 柱平衡缓冲液 A1 (20 mmol/L Tris-HCl,pH 8.0, 150 mmol/L NaCl),4 ℃高压匀浆破碎后,12 000 r/min 离心 30 min 收集上清液, 经 0.22 μm微孔滤膜过滤,缓慢上样于Ni 柱亲和吸附柱,缓冲液 B1 (20 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl,500 mmol/L 咪唑)线性洗脱,紫外检测仪在线检测洗脱峰,收集洗脱液进行SDS-PAGE 电泳检测。
将初步纯化后的样品超滤浓缩,并更换缓冲液A2 (20 mmol/L Tris -HCl, pH 8.0, 200 mmol/L NaCl),上样于 Dextrin Sepharose HP 亲和吸附柱上,用缓冲液A2 洗脱杂蛋白质,缓冲液B2(20 mmol/L Tris-HCl, pH 8.0, 200 mmol/L NaCl,10 mmol/L 麦芽糖) 洗脱目标蛋白质。 收集洗脱液, 用9% SDSPAGE 电泳检测纯化效果。
1.2.6 牛津杯法抑菌活性测定 准备E. coli BL21/pET-MBP-MET 细胞破碎上清液、 纯化后 MBPMET、MBP-proMET 融合蛋白液。 从 E. coli JM109无抗平板上挑取单菌落至5 mL LB 液体培养基,37 ℃培养 4 h, 转接 500 μL 种子液至 50 mL LB 培养基中,37 ℃培养 2 h 至 OD600=0.5。 预先倾倒20 mL LB 培养基至玻璃平板铺平凝固。 吸取1 mL菌液至200 mL 保温于50 ℃的琼脂培养基中混匀,快速吸取5 mL 至平板铺平待凝。 待上层培养基凝固后,将灭菌后的牛津杯(8 mm × 6 mm × 10 mm)垂直立于平板标志附近。 牛津杯中分别加入过滤除菌的 100 μL E. coli BL21/pET-MBP-MET 细胞破碎上清液和纯化后MBP-MET、MBP-proMET 融合蛋白液(0.25 mg/mL),100 μL Amp(0.1 mg/mL)做阳性对照。 盖上平皿盖,将平皿置于37 ℃恒温培养箱培养16~18 h。 观察抑菌圈情况。
1.2.7 最小抑菌浓度(MIC)测定 采用微量稀释法测定MBP-MET、MBP-proMET 融合蛋白质的最低抑菌浓度。 对数生长期的E. coli JM109 菌液按1 :105稀释至 106cfu/mL, 取 100 μL 菌悬液加入 U 型微量板。 过滤除菌后的 MBP-MET、MBP-proMET 稀释至终质量浓度分别为 160、140、120、100、80、60、40、20、10、5 μg/mL, 聚苯乙烯 U 型微量板每孔加入100 μL 稀释后蛋白质液。 对照组加入 100 μL 无菌水,37 ℃振荡培养18~20 h 后观察结果。
2 结果与分析
2.1 met、promet 基因与 mbp 基因的扩增
蜂毒肽met、 前蜂毒肽promet 与麦芽糖结合蛋白 mbp 基因经 PCR 扩增后分别获得 126、252、1 152 bp 的特异性片段, 见图2。 重叠延伸后得到MBP-MET 和 MBP-proMET 片段。 将 PCR 产物连接至克隆载体pMD19-T,测序结果表明MBP-MET 基因片段正确插入pMD19-T 载体。

图2 pET-MBP-MET 表达载体构建Fig. 2 Construction of pET-MBP-MET expression vector
2.2 重组表达载体pET-MBP-MET、pET-MBP-proMET 构建
对 pMD19T-MBP-MET、pET-MBP-proMET 和pET15-b 分别使用限制性内切酶Xho I 与Nco I 进行双酶切处理, 胶回收片段大小为1 300 bp 的MBP-MET 基因片段、1 400 bp 的 MBP-proMET 片段和5 600 bp 的pET15-b 线性片段,使用T4 DNA连接酶过夜连接,连接产物转化E. coli JM109,在氨苄抗性平板上挑取阳性克隆,提取的质粒进行测序鉴定, 得到 6 917 bp 的 pET-MBP-MET 和 7 043 bp的pET-MBP-proMET。
2.3 蜂毒肽融合蛋白质的诱导表达
将表达质粒 pET-MBP-MET 转入 E. coli BL21,挑取单菌落培养,在 0.1 mmol/L IPTG、30 ℃条件下诱导表达,收集细胞,每100 mL 培养基能收集0.6 g 湿菌。 重悬并超声破碎后离心收集上清液,进行SDS-PAGE 验证。 结果如图3 所示,上清液可见相对分子质量 46 200(见图 3(a))和 50 800(见图3 (b)) 的条带, 分别与融合蛋白质 MBP-MET 和MBP-proMET 理论大小一致,且上清液中目标条带浓度高于沉淀。 经凝胶电泳成像仪分析,上清液的目标条带占全细胞总量的15%, 说明融合蛋白质MBP-MET 在重组大肠杆菌中主要以可溶性蛋白质的形式表达。

图 3 E. coli BL21/pET-MBP-MET、E. coli BL21/pETMBP-proMET 表达产物SDS-PAGEFig. 3 SDS-PAGE analysis of expression products of E. coli BL21/pET-MBP-MET and E. coli BL21/pET-MBP-proMET
2.4 融合蛋白质MBP-MET 和MBP-proMET 的纯化
将诱导表达后的重组菌E. coli BL21/pETMBP-MET 和 E. coli BL21/pET-MBP-proMET 分别离心收集菌体, 加入缓冲液A1 (20 mmol/L Tris -HCl, pH 8.0, 150 mmol/L NaCl) 重悬后用高压匀浆机破碎, 离心后的上清液经0.22 μm 微孔滤膜过滤,使用Ni 柱亲和吸附柱纯化,分别用含30 mmol/L咪唑和100 mmol/L 咪唑的缓冲液B1 洗脱, 紫外检测到出现洗脱峰。 收集洗脱液进行SDS-PAGE 电泳检测,结果见图4。 30 mmol/L 咪唑洗脱大量杂蛋白质,100 mmol/L 咪唑洗脱时目标蛋白质几乎全部洗脱下来。 但经过Ni 柱亲和柱纯化后的目标蛋白质里含有杂质,需经过进一步纯化。 因此将初步纯化后的样品超滤浓缩, 并更换平衡缓冲A2(20 mmol/L Tris - HCl, pH 8.0, 200 mmol/L NaCl),上样于Dextrin Sepharose HP 亲和吸附柱上, 用缓冲液A2 洗脱杂蛋白质, 以10 mmol/L 麦芽糖的缓冲液B2 洗脱目标蛋白质,SDS-PAGE 结果见图 5。 经凝胶电泳成像仪分析SDS-PAGE 结果,最终得到纯度高达90%的蛋白质样品。 通过计算,每升菌液可以纯化得到20 mg 左右的纯蛋白质。

图 5 融合蛋白质 MBP-MET、MBP-proMET 的 Dextrin Sepharose HP 纯化Fig. 5 Purification of fusion protein MBP -MET and MBP-proMET with Dextrin Sepharose HP
2.5 牛津杯法测定MBP-MET 和MBP-proMET的活性
通过牛津杯实验鉴定融合蛋白质MBP-MET 和MBP-proMET 的抑菌活性。 从图6 可知,与阴性对照相比,纯化后的MBP-MET 和MBP-proMET 均对大肠杆菌有明显的抑制效果。 经测量MBP-MET 的抑菌圈直径为 9.2 mm(见图 6(a)),MBP-proMET 的抑菌圈直径为 8.8 mm(见图 6(b))。MBP-MET 的抑菌效果比MBP-proMET 好。可能由于融合蛋白质的相对分子质量比较大且结构较抗生素复杂,并且实现其生物活性还需要其特定的空间结构,而琼脂平板的网状结构不利于融合蛋白质快速的扩散到周围,使得其抑菌圈相对抗生素的较小。
2.6 MBP-MET 和MBP-proMET 的最低抑菌浓度
微量稀释法判断不同质量浓度MBP-MET 和MBP-proMET 的纯化后蛋白质对E. coli JM109 的抑菌效果,对照组有细菌生长,将肉眼观察到没有细菌生长的培养孔中的药物质量浓度定为MIC,判断生长抑制标准为肉眼观察未发现培养液混浊或沉淀现象出现。 从表 2 可知,MBP-MET 对 E. coli JM109 的 MIC 为 100 μg/mL,MBP-proMET 对 E. coli JM109 的 MIC 为 140 μg/mL。

图 6 融合蛋白质 MBP-MET、MBP-proMET 对 E. coli JM109 的抑菌效果Fig. 6 Antibacterial activity of fusion protein MBP-MET and MBP-proMET on E. coli JM109
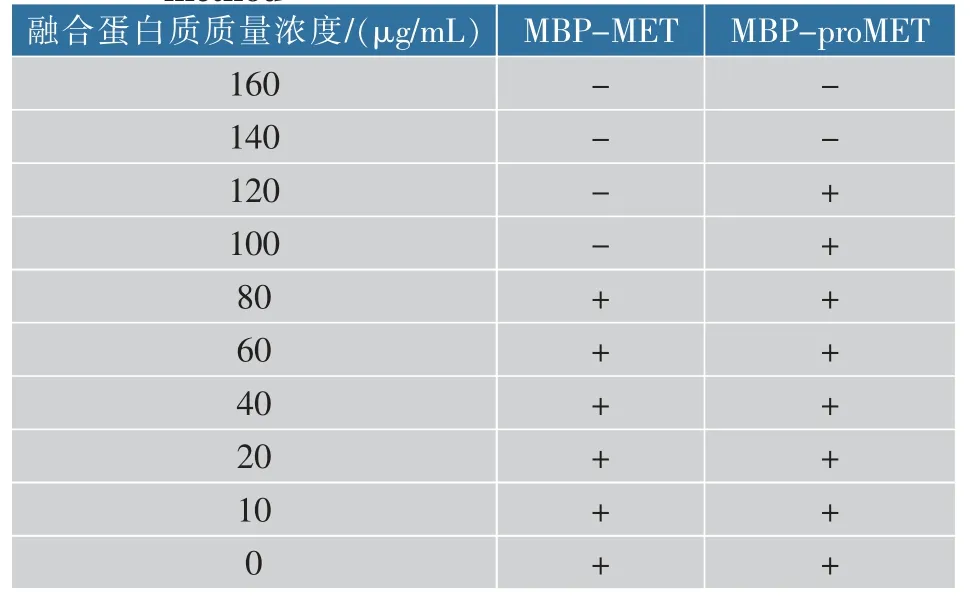
表2 微量稀释法测定MBP-MET、MBP-proMET 对大肠杆菌的MICTable 2 Determination of the MIC of MBP-MET and MBP-proMET against E. coli by microdilution method
3 结 语
蜂毒肽作为一种具有抗菌、抗炎、抗肿瘤、溶血等多种生理活性的活性肽,已受到广泛关注。 但由于蜂毒肽对细胞具有一定的致死性,蜂毒肽在原核生物和真核生物中难以表达。
人们曾通过重组蜂毒肽与其它功能性基因融合表达。文良柱等[22]将蜂毒肽5~12 位氨基酸与死亡素4~21 位氨基酸基因克隆在载体pET32-a 上,在E.coli BL21(DE3)中融合表达成杂合肽MT,但目标肽的产率没有明确。 张宏刚等[23]将蜂毒肽(3~14)与贻贝素B(13~27)杂合,在巴斯德毕赤酵母(Pichia pasioris)中通过甲醇诱导表达获得Mel - MytB 杂合肽,直接进行表达产物的抑菌分析,结果得到表达产物对大肠杆菌、金黄色葡萄球菌、鸡沙门氏菌等具有抑菌活性,但该实验并未对表达产物进行纯化得到单纯杂合肽,且诱导时间长达24~72 h,培养时间较长。 杨光勇[24]通过SOE-PCR 技术连接抗白细胞介素 4 受体 (IL-4R) 单链抗体 scFv 与蜂毒肽(MLT)基因,构建抗IL-4R 单链抗体连接蜂毒肽的重组质粒 pET-32a-scFv-MLT, 转入 E.coli BL21(DE3)内诱导表达,scFv-MLT 融合蛋白质主要以包涵体形式表达。
在重组蜂毒肽的文献报道中,标签蛋白质谷胱甘肽转移酶 (GST) 在与蜂毒肽重组表达中应用最多,但GST 在增加蜂毒肽的可溶性表达上存在一定局限。 胡宗利等[25]将蜂毒肽(melittin)基因序列定向克隆到表达载体pESP-2 中, 构建GST-melittin 融合蛋白质,在裂殖酵母(Schizosaccharomyces pombe)中进行异源表达,1 L 培养液收集到约5 g 鲜活酵母菌,纯化得到400 μg 融合蛋白质,存在诱导时间较长,目标肽产率相对较低等局限。朱文赫等[26]将人工合成整合了肠激酶作用位点的蜂毒肽 (MEL)基因, 插入到载体pGEX-2T, 构建融合表达载体pGEX-MEL, 通过 E. coli BL21 (DE3) 成功表达GST-MEL 融合蛋白质,但可溶性表达产物较少,每毫克融合蛋白质经肠激酶切割后得到78.8 μg 蜂毒肽。 Buhrman 和 Jason S[27]构建了含有 GST-6×Hismelittin 的质粒 pJB-HTS-melittin 并在 E. coli Rosetta 2 细胞里表达,并用不断增加浓度的轻度非离子去污剂从不溶部分重复萃取, 富集了GST-6×His-melittin 融合蛋白质,每升菌液能得到0.5~1 mg蜂毒肽融合蛋白。
作者采用融合表达的方案,首次应用麦芽糖结合蛋白质作为融合伴侣,通过增加溶解性增加了蜂毒肽的表达量,同时构建了含信号肽和前导肽的前蜂毒肽重组菌株, 克服了蜂毒肽对细胞的毒性,实现了蜂毒肽的可溶性表达。 通过Ni 柱和Dextrin Sepharose HP 两步亲和纯化, 得到了高纯度的融合蛋白质MBP-MET 和MBP-proMET。 牛津杯抑菌实验鉴定了两种融合蛋白质的抑菌活性, 且MBP-proMET 对大肠杆菌的最低抑菌浓度高于MBPMET,猜测可能是由于引入的前导肽和信号肽结构域中带有高度负电荷增加了分子整体所带负电荷,因而比蜂毒肽成熟肽单独表达具有更大的溶解度,因而降低了对细胞膜的损伤能力。 最低抑菌浓度高于 Tony Picoli 等[28]报道的 42.5 μg/mL,说明麦芽糖融合蛋白质在增加蜂毒肽的溶解性的同时,其大分子结构对抑菌效果有一定的影响。 在试验探究过程中, 曾设计采用TEV 蛋白酶切割大分子融合标签,发现MBP 标签蛋白质不完全影响蜂毒肽的功能,但对TEV 酶的切割有较大影响,究其原因可能是标签蛋白质部分包裹了蛋白酶切位点。 也尝试将蜂毒肽基因构建在pGEX-6p-1 上进行原核表达, 但目标蛋白质GST-MET 主要以包涵体形式表达, 说明不同的融合伴侣蛋白质对蜂毒肽的异源表达有较大的影响。蜂毒肽的蛋白质结构已经获得解析[13],整体结构为一个完整的α 螺旋,在后续研究中,我们将尝试GST-MET 包涵体变复性实验以及纯化,并采用圆二色谱对MET 的二级结构进行复性监控,进一步为“纯净”蜂毒肽的制备奠定较坚实的研究基础。
