维生素K2固体自乳化制剂的处方优化
2020-11-09赵运双金小红梅兴国
赵运双,陈 洁,金小红*,梅兴国
(1.湖北科技学院药学院,湖北 咸宁 437100;2.军事科学院军事医学研究院,北京 100850)
固体自乳化药物传递系统(solid self-microemulsifying drug delivery system,SSEDDS)是由药物、油相、非离子型表面活性剂、助表面活性剂与适宜的固体材料混合制备成的固体制剂[1],并能在胃肠道内或在环境温度(通常指体温37℃)及温和搅拌的情况下能自发形成水包油乳剂。与传统自乳化药物传递系统(self-microemulsifying drug delivery system,SEDDS)相比,SSEDDS稳定性增加、贮存时间延长、胃肠道刺激性减少、服用方便。因为其能够提高脂溶性或水难溶性药物的溶解度、生物利用度以及降低胃肠道副作用,所以常用于运送难溶性药物[2]。
维生素K2(vitamin K2,VK2),也称甲基萘醌(menaquinone,MK),作用于肝外组织(骨和血管等器官),将无机钙与有机蛋白结合成骨基质和阻止血管钙化。现在市面上有多种VK2类药物上市,主要剂型为软胶囊。但由于VK2在水中的溶解度较低,使其难以通过小肠绒毛表面的亲水层到达小肠上皮细胞[3]。本研究借助有效的制剂手段,将VK2开发为口服自乳化制剂达到提高口服VK2生物利用度的目的。
1 材料与仪器
1.1 材料
VK2原料药(含量质量分数0.2%,广东双骏生物科技有限公司),油酸(上海麦克林生化科技有限公司),油酸乙酯(山东瑞生药用辅料有限公司),中链甘油三酯(新兴铁岭药业股份有限公司),大豆油(山东瑞生药用辅料有限公司),吐温80(天津市科密欧化学试剂有限公司),吐温20(天津市科密欧化学试剂有限公司),聚氧乙烯40氢化蓖麻油(德国BASF公司),聚乙二醇400(天津市博迪化工有限公司),甘油(中国医药集团上海化学试剂公司),二乙二醇单乙醚(上海麦克林生化科技有限公司),异丙醇(常熟市鸿盛精细化工有限公司),微晶纤维素(湖北兴银河化工有限公司),糊精(湖北兴银河化工有限公司),淀粉(山东萍聚生物科技有限公司),乳糖(河南圣茂精细化工有限公司),聚乙二醇6000(江苏梦得新材料科技有限公司),麦芽糊精(湖北裕盈生物科技有限公司),水溶性淀粉(湖北裕盈生物科技有限公司),甘露醇(河北步润生物科技有限公司),蒸馏水(自制)。
1.2 仪器
RCZ-6B3溶出度测试仪(上海黄海药检),Agilent1100系列高效液相色谱仪(美国安捷伦公司),LD5-2B高速离心机(北京雷勃医疗器械),DF-101S集热式恒温加热磁力搅拌器(巩义市予化仪器有限责任公司),QL-901涡旋仪(海门市其林贝尔仪器制造有限公司),KQ-250DV型数控超声清洗仪(昆山市超声仪器有限公司),SHZ-82A恒温水浴震荡器(金坛市医疗仪器振荡器),FA2104B电子分析天平(上海精密仪器仪表有限公司),纳米粒度及电位分析仪(美国PSS公司),WBF-ZG多功能流化床(重庆英格造粒包衣技术有限公司)。
2 方法与结果
2.1 基本辅料的溶解度试验
在预试验基础上选取可形成自乳化的油相、乳化剂、助乳化剂进行研究。分别置10mL试管中,加入过量VK2原料药,超声溶解30min,观察溶解情况,直至饱和。室温25℃,水浴震荡24h,离心后取上清液,过滤注入液相色谱仪,记录色谱图。按外标法以峰面积计算含量。
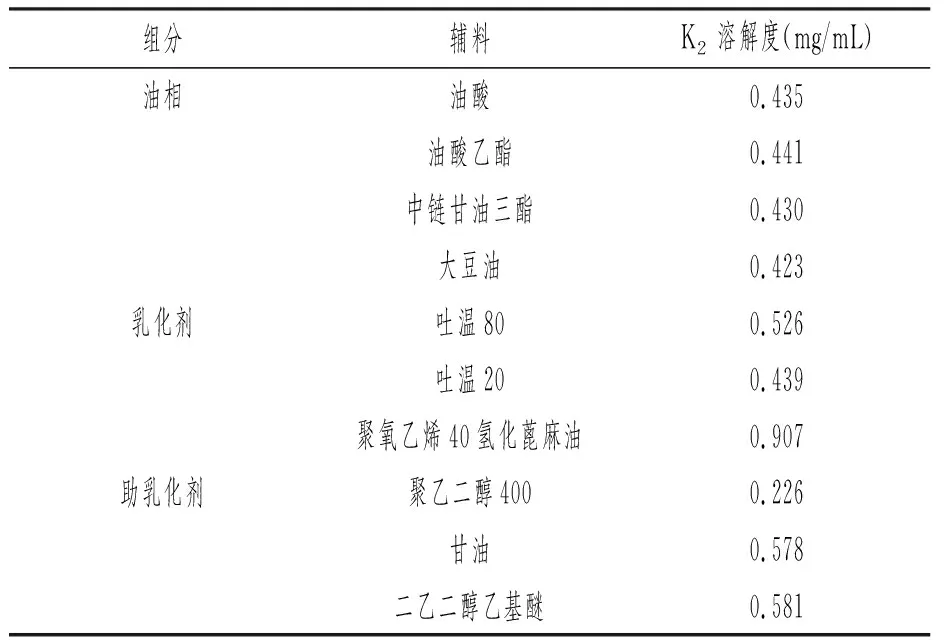
表1 VK2在不同辅料中溶解度
2.2 处方配伍筛选
选择油酸、油酸乙酯、中链甘油三酯、大豆油为油相,溶解性较好的聚氧乙烯40氢化蓖麻油为乳化剂,进行配伍筛选,结果见表2。称取不同比例的处方混合均匀,取1g混合物加入到100mL 37℃的蒸馏水中,用磁力搅拌器温和搅拌,目测自乳化效果。将自乳化效果分为5个级别[4-5]:A级(乳化时间<1min,溶液澄清,透明或微泛蓝光);B级(乳化时间<1min,溶液呈半透明,呈蓝白色);C级(乳化时间<2min,呈亮白色不透明液体);D级(乳化时间>2min,色泽暗,呈灰色,略带油状);E级(难乳化,一直有油滴存在)。

表2 不同油相和乳化剂配伍试验结果
备注:Ⅰ聚氧乙烯40氢化蓖麻油-油酸;Ⅱ聚氧乙烯40氢化蓖麻油-油酸乙酯;Ⅲ聚氧乙烯40氢化蓖麻油-中链甘油三酯;Ⅳ聚氧乙烯40氢化蓖麻油-大豆油
由表2可知油酸乙酯与聚氧乙烯40氢化蓖麻油配伍相容性较好。所以油相选用油酸乙酯,乳化剂选用聚氧乙烯40氢化蓖麻油。因为甘油对聚氧乙烯40氢化蓖麻油有增加增溶作用,且二乙二醇乙基醚价格较贵,所以助乳化剂选用甘油。
2.3 三元相图的绘制
按乳化剂∶助乳化剂1∶3、1∶5、1∶7、1∶9、1∶1、9∶1、7∶1、5∶1、3∶1,油相所占比例为12.5%、25.0%、50.0%、62.5%、75.0%、87.5%进行自乳化效果考察。以油酸乙酯、聚氧乙烯40氢化蓖麻油、甘油为三元相图3个顶点,用绘图软件Origin 8作图1。
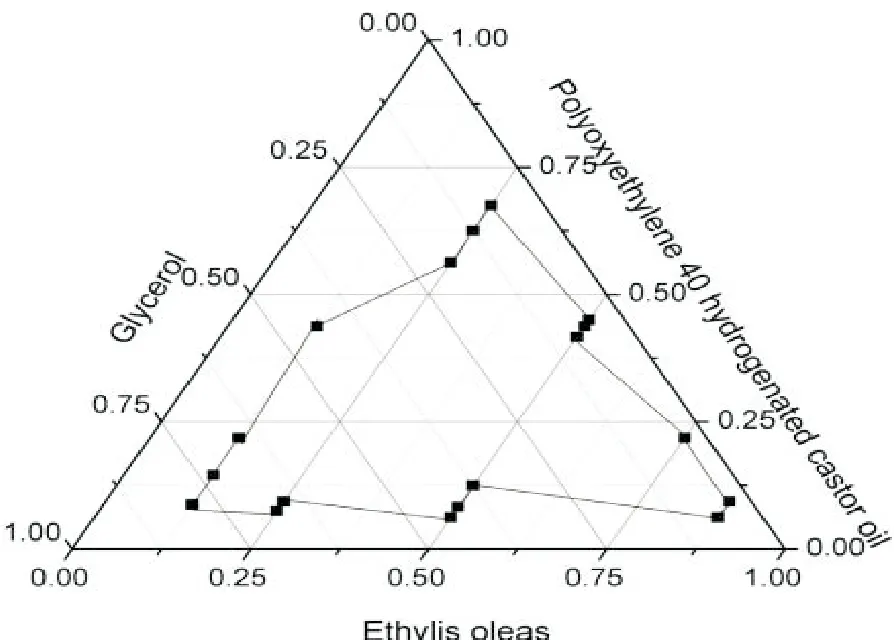
图1 三元相图
2.4 处方优化
2.4.1 实验设计
在绘制三相图的基础上,将含油量(X1,%)、表面活性剂与助表面活性剂的比例Km(X2)作为星点设计-效应面优化法的两个考察因素。根据自乳化体系评判标准,选取自乳化效果较好区域,X1取值范围为25%~40%,X2取值范围为1~3。按照星点设计的设计原理,采用Design-Expert 8.05软件进行二因素五水平(-a、-1、0、1、a)试验设计[6-8]。各因素水平代码如表3所示。制备自乳化制剂,以平均粒径,Zeta电位和多分散系数作为稀释后所形成乳剂的三个评价指标,共计13个处方进行系列指标测定。

表3 星点设计的因素水平及代码
2.4.2 测定电位、粒径、分散系数
按照星点设计方案,称取处方量的辅料,混合均匀,配制13组空白自乳化组份,分别称取4g置溶出仪,温度设为37℃,400mL蒸馏水稀释100倍,75r/min搅拌均匀后取适量,使用粒径测定仪分别测定自乳化液体的粒径、Zeta电位和分散系数,见表4。
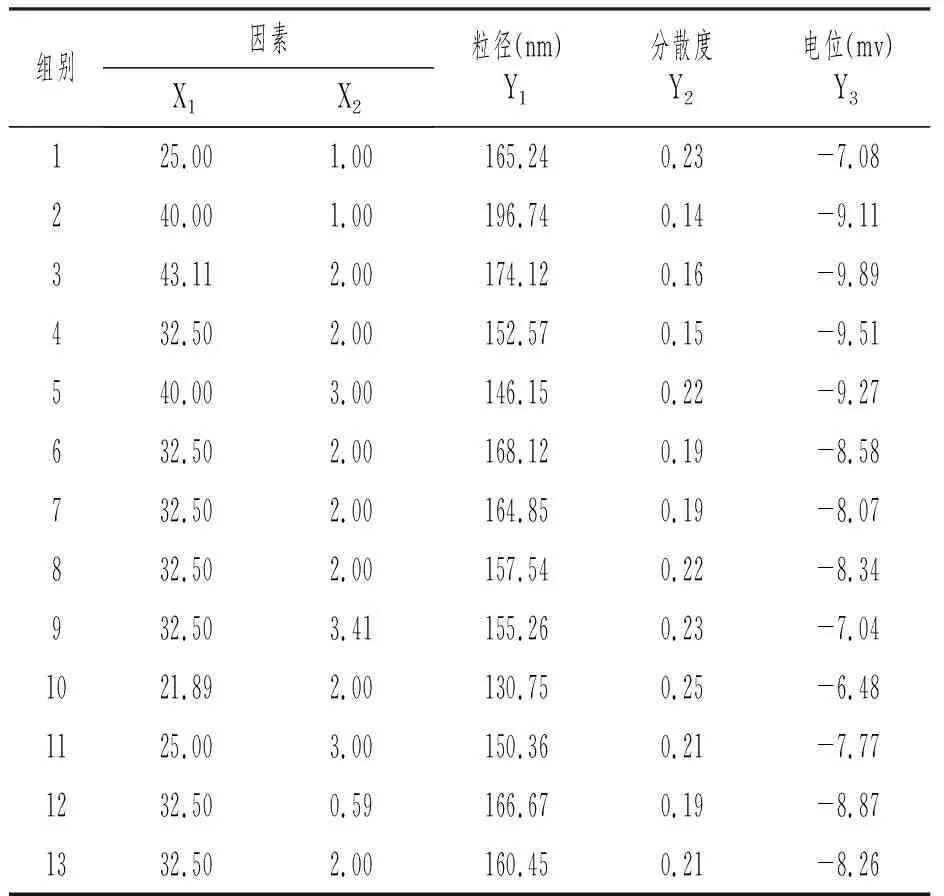
表4 星点设计表及响应值
粒径=+23.70771+6.3478X1+20.04456X2-1.19033X1X2-0.038309X12+2.11012X22(R2=0.5768,P=0.0422<0.05)
分散系数=+0.60715-0.015466X1-0.12226X2+3.333×10-3X1X2+8.222×10-5X12+7.125×10-3X22(R2=0.5778,P=0.0419<0.05)
电位=-0.026624-0.31797X1-1.31342X2+0.01766X1X2+2.20667×10-3X1+0.23913X22(R2=0.5845,P=0.0398<0.05)
由上述方程可知,结果具有统计学意义。
采用Design Expert 8.05进行拟合作图,得效应面曲线(见图2~4,封二)。可直观反映各因素和响应值及各考察因素之间的交互作用。
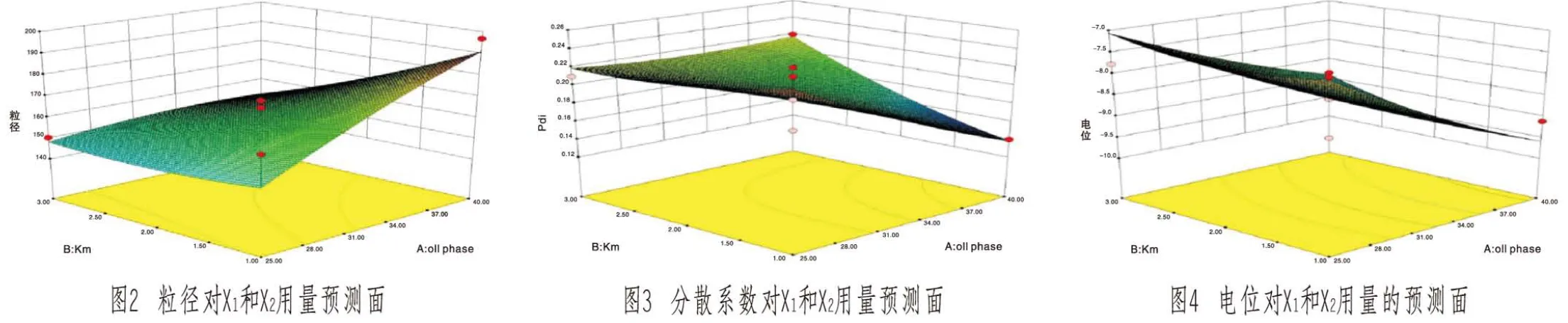
根据各效应面的三维图及模型拟合得出的方程,运用Design Expert 8.05的预测功能得出因素X1、X2的最终取值,即X1=40%,X2=2.07,得出VK2自乳化的最优处方为油酸乙酯40%,聚氧乙烯40氢化蓖麻油40.5%,甘油19.5%。
2.5 优化处方的验证
根据预测的优化处方制备空白自乳化制剂,分别测定粒径、电位和分散系数,将实测值与预测值作比较(见表5)。结果表明,采用的星点设计-效应面法具有较高的预测准确性。
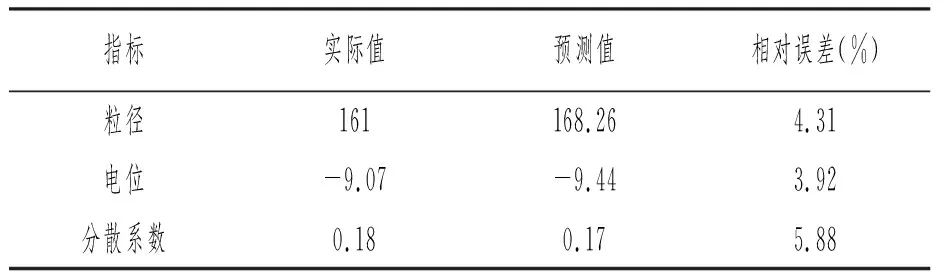
表5 预测值与实际值(n=3)
2.6 VK2在自乳化制剂中的溶解度
按最优处方制备空白自乳化制剂,将空白自乳化制剂称取5g至离心管中,加入过量VK2。超声后放于恒温水浴震荡床中,震荡24h后以8000r/min离心转速离心10min,用移液枪移取1mL上清液至10mL容量瓶中,用异丙醇稀释至刻度。用外标法进行HPLC检测,测得VK2在自乳化制剂中平衡溶解度为1.463mg/mL。
2.7 加药量
按优化处方称量油酸乙酯、聚氧乙烯40氢化蓖麻油和甘油,于37℃水浴中超声,使其混合均匀,加入占处方量10%、30%、50%、70%的VK2原料药超声至完全溶解,将其加入到100倍37℃保温的蒸馏水,75r/min搅拌下自乳化。测定粒径和乳化时间,选择合适的加药量。结果如表6所示。
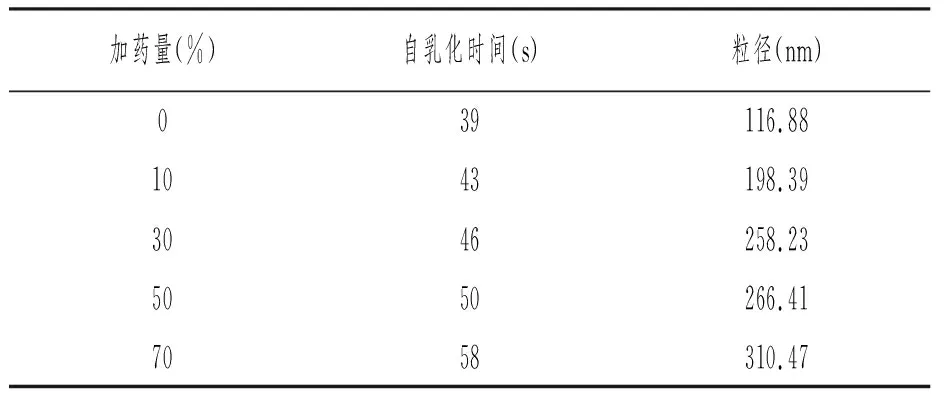
表6 加药量对自乳化速率影响
含药自乳化制剂乳化所需时间和乳化后粒径较空白处方有增加,但粒径均保持在500nm以下,乳化时间均小于60s,表明在处方对VK2的饱和溶解度范围内,加药量对自微乳乳化效率影响不大,但是随着载药量的进一步增大,体系澄明度下降。且当加药量为70%时,已接近处方的饱和溶解度,考虑到自乳化制剂贮存过程中可能会存在药物析出问题,且K2原料药中的辅料对自乳化效果的影响,加药量离饱和溶解度应留有一定余地,故加药量定为50%(1g原料药中含纯K22mg)。
2.8 固体吸附剂筛选
称取1g固体粉末于10mL小烧杯,往里面滴加自乳化稀释后液体,直至粉体表面无可见液滴,记录此时自乳化液体所用量。如表7所示。
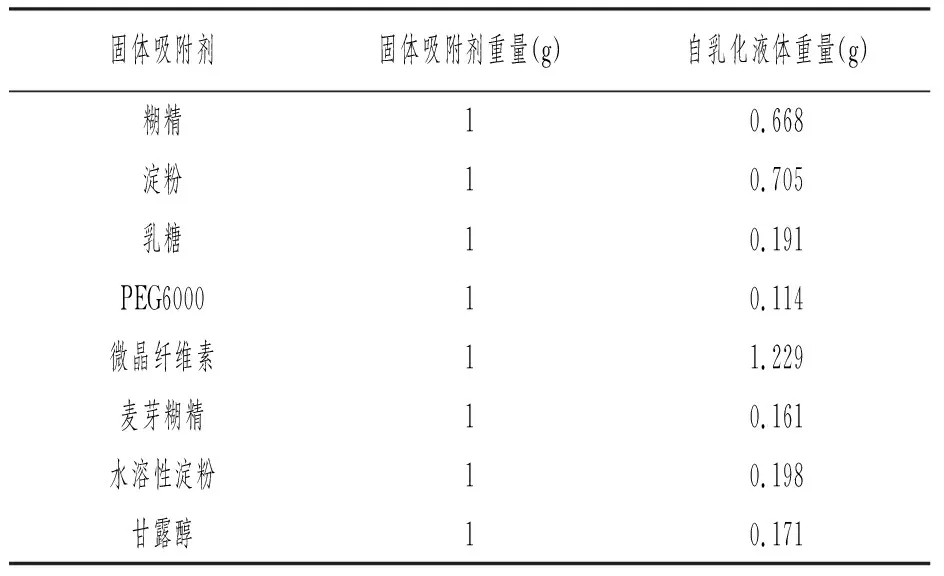
表7 固体载体吸附度
由表7可知水不溶性材料的吸附能力强于水溶性吸附材料,微晶纤维素属于水不溶性材料。微晶纤维素价格低廉且不易与主药发生反应,是制药工业中液体固化时常用到的吸附剂,与淀粉类、乳糖相比,微晶纤维素具有较大的比表面积,因此,较少量便能使液体药物转化成无粘附、干燥的液固粉末。因为微晶纤维素的吸附性能较好,且吸附后对自乳化效果影响较小,所以选用微晶纤维素对自乳化液体进行固化。
2.9 固体载体与VK2 SEDDS的比例选择
称取6份含药SEDDS各30g,分别加300mL纯化水,室温搅拌状态下,得到均匀乳剂,然后分别加入不同量的微晶纤维素30g,60g,90g,120g,150g,180g,使之比例分别为1∶1,2∶1,3∶1,4∶1,5∶1,6∶1搅拌混匀。
WBF-2G流化床的设置参数为:进风温度115℃,转速为5r/min,流速30Hz,出风温度为80℃。按上述方法干燥喷雾,考察喷雾干燥的情况。结果见表8。

表8 固体载体微晶纤维素与VK2 SEDDS的比例选择结果
选取固体载体与自乳化载药体系为4∶1的比例,并通过喷雾干燥得到流动性较好的固体自乳化粉末。
2.10 溶出度
按照《中国药典》附录有关浆法规定进行[9],水浴温度(37±0.5)℃,保温搅拌状态下,溶出介质为pH 6.8磷酸缓冲溶液500mL。分别于5、10、15、20、30、60min取样5mL,同时补加5mL同温度的同种溶出介质,经0.22μm微孔滤膜过滤,取50μL注入高效液相色谱仪,记录峰面积,用标准曲线法计算累积溶出百分率,并以VK2原料药作为对照。如图5所示。

图5 平均累积溶出率与时间的关系
由图5可知,VK2自乳化粉末在pH 6.8缓冲液中有较好的溶出效果,在30min时,溶出度达到90%以上,基本完全溶出。VK2原料药在pH6.8介质中几乎不溶。与原料药溶出度相比VK2自乳化粉末溶出速度显著增加。
3 讨 论
选用水不溶性载体微晶纤维素为固体载体,以喷雾干燥法制备VK2自乳化固体分散体。当VK2自乳化制剂∶微晶纤维素(w/w)=1∶4时,制备的固体分散体外观较好,且有较好的乳化效果。
本实验以Ethyl Oleate为油相,RH40为表面活性剂,Glycerol为助表面活性剂,评价体系的自乳化速率,并进行了质量评价。选用星点设计-效应面对VK2进行处方优化,经验证,该优化处方平均粒径、电位和分散度实际值与理论值吻合良好,符合设计要求。
VK2在人工肠液中几乎不溶,制成自乳化固体粉末后,其溶解性显著提高,累计溶出百分率在5min时达60.89%,30min达91.48%,溶出明显优于VK2原料药。
