电感耦合等离子体原子发射光谱法测定铀氧化物中微量碱金属元素
2020-10-28陈岚,徐鸿,邵燕
陈 岚,徐 鸿,邵 燕
中核建中核燃料元件有限公司,四川 宜宾 644000
在核燃料领域中,铀氧化物应用较为广泛,其中二氧化铀芯块以其独特的性能优点几乎成为所有轻水堆的主要燃料[1]。燃料芯块作为核燃料元件的核心部分,制造过程中对核纯级二氧化铀粉末、芯块产品中化学要求、总硼当量等指标进行了严格的规定[2],碱金属元素作为芯块产品中的杂质元素以及用于计算总当量硼含量所需的特定元素,不仅规定了单个元素的最高含量限值而且规定了杂质元素总含量限值,因此需要对Li、Na、K、Cs等碱金属元素进行准确的检测,以满足核燃料产品的制造要求。
国内常用空气-乙炔原子吸收分光光度法(AAS)/火焰发射光谱法(FAES)[3]联合分析二氧化铀芯块中的碱金属元素,该分析方法准确度高,但使用空心阴极灯作为气体放电光源,只能进行单元素的测定,效率相对较低,且灯的维护保养要求较高;同时使用乙炔气体作为燃气,对安全要求较为严格。
电感耦合等离子体原子发射光谱法(inductively coupled plasma atomic emission spectrometry, ICP-AES)被公认为是最有前途的常规分析工具之一,与原子吸收光谱法相比,其具有对痕量和微量元素的测定分析灵敏度高、稳定性好、线型范围宽、受基体干扰小、多元素同时测定分析速度快的优点。美国材料与试验协会已建立ICP-AES测定铀氧化物中67种杂质元素的分析标准[4]。本工作拟通过选择合适的分析条件减少碱金属类易电离元素所造成的电离干扰[5-6],建立等离子体发射光谱法同时测定铀氧化物中微量Li、Na、K、Cs的分析方法,以提高分析效率。
方法有效性验证拟采用标准加入法进行回收实验和方法比对两种方式,鉴于目前尚没有以铀为基体的碱金属元素有证标准物质,比对样品制备根据GB/T 13370-92[3]中元素的测量范围,制备两个含量水平的工作标准物质,元素含量值分别接近于GB/T 13370-92分析方法中规定的元素测量范围上限值和下限值。
1 实验部分
1.1 仪器与试剂
OPTIM4300DV型全谱直读电感耦合等离子体发射光谱仪,美国PE公司;ES225SM-DR分析天平,精度0.1 mg,美国普利赛斯公司;HT-200可调温电热板,广州格丹纳公司。
100 mg/L Li、Na、K单元素标准贮备溶液和1 000 mg/L Cs单元素标准贮备溶液,北京化工冶金研究院;CL-TBP萃淋树脂,萃取剂(磷酸三丁酯)质量分数为60%,粒径为0.125~0.193 mm,常州大学;石英萃取色层柱,自制,内径8 mm,柱体长180 mm,上方带约30 mL储液斗。
硝酸、盐酸等,市售优级纯;实验用水为去离子水,自制。
1.2 仪器工作条件
高频发生器功率950 W,等离子气流量15 L/min,雾化气流量1.0 L/min,辅助气流量0.2 L/min,溶液提升率2.0 mL/min,最大读数时间为5 s,轴向(水平)观测方式。分析波长Li 670.784 nm、Na 589.592 nm、K 766.490 nm、Cs 455.530 nm。
1.3 实验方法
1.3.1萃取色层柱的制备 将一定量的CL-TBP萃淋树脂用水浸泡至少24 h,湿法装入萃取色层柱,树脂床高约150~160 mm,控制流速为0.5~1.2 mL/min,色层柱内树脂床的上下两端均用聚四氯乙烯丝或脱脂棉填塞。装好的柱子用5.5 mol/L硝酸溶液和浓盐酸溶液反复交替淋洗至淋出液的杂质元素含量小于分析方法下限,用3.0 mol/L硝酸溶液平衡色层柱。分离后,用40 mL去离子水解吸色层柱上吸附的铀,然后将萃取色层柱浸泡在去离子水中重复使用。
1.3.2样品处理 样品溶解:称取0.500 0 g铀氧化物样品置于25 mL石英烧杯中,加入2~4 mL浓硝酸,于电炉上低温加热,保持微沸状态,待红棕色烟雾消失溶液呈澄清的黄绿色溶液,此时样品溶解完全,继续将溶液加热蒸干成橘红色固体干渣,然后加入2 mL 3.0 mol/L硝酸溶液溶解干渣。分离铀基体:将2 mL样品溶液全部转移至平衡好的萃取色层柱上,继续用10 mL 3.0 mol/L硝酸溶液分多次淋洗烧杯和色层柱,收集第3—12 mL淋出液共10 mL于容量瓶中,得待测样品溶液A1。空白样品:在不加样品的情况下,重复上述步骤,得试剂空白A0。
1.3.3样品测定 在选定的仪器工作条件下,将混合标准溶液系列依次引入ICP光源,测定不同浓度标准溶液的仪器响应信号,由仪器计算机自动绘制出标准曲线,要求测得的各元素标准曲线线性相关系数不小于0.99;然后依次将试剂空白A0及样品溶液A1引入ICP光源进行分析测定。
2 结果与讨论
2.1 工作条件的选择
碱金属元素属于低电离电位易激发的元素,元素谱线分布在较低观测高度。谱线峰值观测高度与炬管结构、高频发射功率以及载气流量有关。实验发现,采用径向(垂直)信号观测方式,ICP光谱仪对溶液中的Li、Na、K元素具有非常好的检测灵敏度,即使在很低的浓度下仪器也有较强的响应信号;但Cs元素的响应信号则较弱,背景偏高,净强度低。为避免观测高度对测量结果的影响,选择轴向(水平)信号观测方式,分别考察了不同高频发射功率、溶液提升率及雾化气流量下仪器对Cs元素的信号响应情况,结果示于图1。图1结果表明,在轴向(水平)信号观测条件下,高频发射功率950 W、溶液提升率2.0 mL/min、雾化气流量1.0 L/min仪器工作条件能获得碱金属元素较好的信背比。
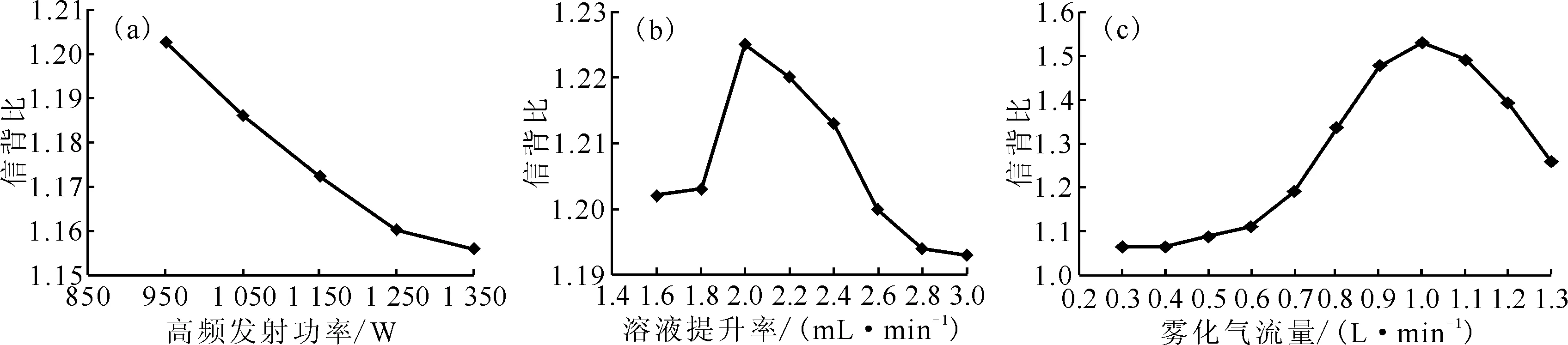
(a)——高频发射功率,(b)——溶液提升率,(c)——雾化气流量
2.2 分析线波长的选择
依据灵敏度高、干扰小、峰型规则、背景平滑的原则,从仪器谱线库中选择Li、Na、K、Cs元素的不同分析线波长,通过核纯级铀氧化物中可能存在的杂质元素及基体铀元素共存条件的实验,选择了与共存元素无光谱干扰的分析线波长:Li 670.784 nm、Na 589.592 nm、K 766.490 nm、Cs 455.530 nm。
2.3 积分算法的选择
尚传胜等[7]研究了用ICP-AES法测定卤水中Rb,发现峰高法相对峰面积法具有更低的检出限。由于ICP光谱仪对Cs元素的响应信号相对较低,容易出现测量灵敏度低、精密度较差的情况,造成检测下限偏高。考虑到Cs与Rb都是碱族元素,在周期表上位置相邻,性质相近。因此,本实验在仪器工作参数下,依次测定混合标准溶液系列,并将2 mg/L Cs元素标准溶液作为样品溶液连续测量10次,分别用峰高法和峰面积法两种积分算法得出各元素的含量及标准偏差,结果列于表1。表1数据表明,Cs元素的测定采用峰高法能有效提高检测灵敏度和精密度,测量结果准确度较高。

表1 不同积分方法测定Cs含量结果的比较
2.4 分离酸度和接收体积
分别使用3.0 mol/L硝酸溶液和5.5 mol/L硝酸溶液作为流动相分离铀基体,按实验方法,每次只接收2 mL淋出液,测定Li、Na、K、Cs各元素的响应信号值绘制成淋洗曲线,结果示于图2。分离完成后,测得铀色层高度分别为9.0 cm和7.5 cm,均能满足分离铀基体的要求,但考虑到酸基体效应和盐基体效应的干扰,尽量降低仪器测量时酸度,选择3.0 mol/L硝酸为分离酸度。从图2可以看出,在不同的分离酸度条件下,碱金属元素均集中在第4—10 mL淋洗液中,为确保杂质收集完全,选择收集第3—12 mL淋洗液作为测量接收体积。

淋洗液c(HNO3),mol/L:(a)——5.5,(b)——3.0
2.5 标准曲线与工作曲线的比较与检验
分别移取Li、Na、K单元素标准贮备溶液(100 mg/L)、Cs元素标准贮备溶液(1 000 mg/L)各1 mL于100 mL容量瓶中,用3.0 mol/L硝酸溶液定容混匀后再逐级稀释得混合标准溶液系列。Li、Na、K元素质量浓度梯度为:0.02、0.05、0.10、0.20、0.50、1.00 mg/L;Cs元素质量浓度梯度为:2、5、10、20、50、100 mg/L。在仪器工作条件下测定,以元素质量浓度为横坐标,信号强度为纵坐标,绘制标准曲线。同时称取0.5 g核纯级 UO2粉末样品,分别加入标准溶液(Li、Na、K:50 mg/L,Cs:1 000 mg/L)0.0、0.4、1.0、2.0、4.0、10.0 mL,按实验方法测定各元素响应信号值,绘制工作曲线。工作曲线和标准曲线的对比示于图3。由图3看出,工作曲线和标准曲线几乎重合。
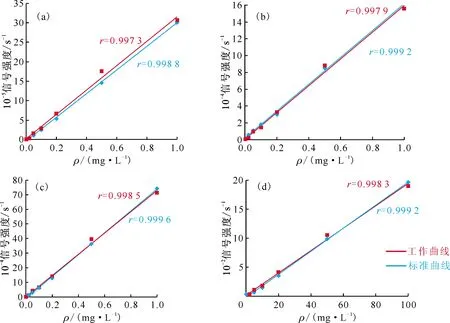
(a)——Li,(b)——Na,(c)——K,(d)——Cs
以F检验比较两条曲线回归标准偏差,以t检验比较两条曲线的斜率,以F检验两条曲线是否重合。曲线的响应信号值和检验结果列于表2。表2结果表明,当置信度为95%时,标准曲线与工作曲线无显著性差异。因此,分析样品时可用标准曲线代替工作曲线。
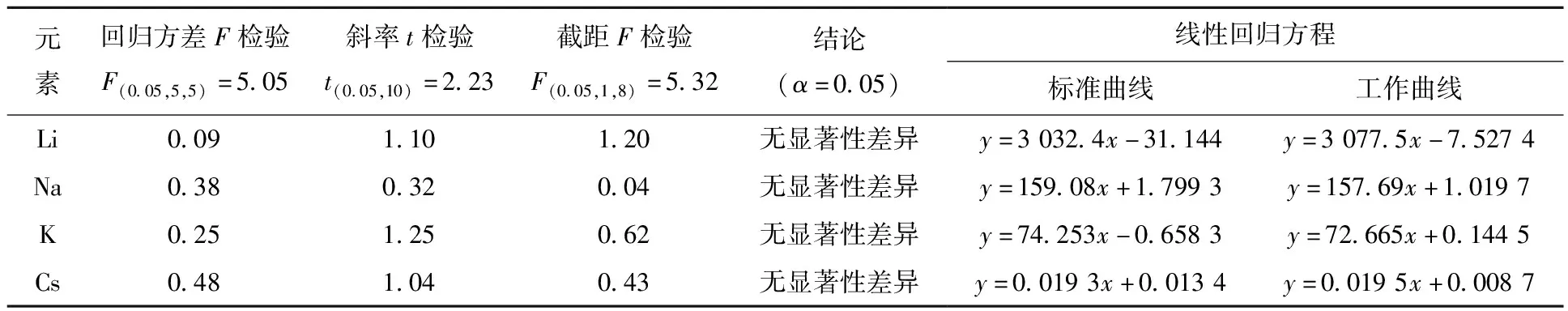
表2 标准曲线与工作曲线的显著性差异
2.6 方法定量检测下限
按照国际纯粹与应用化学联合会(IUPCA)的建议[8],本方法选择不含待测元素的二氧化铀粉末作为空白样品,分三次测定20个空白样品中各元素的含量,结果列于表3。按 10倍标准偏差所对应的浓度计算方法的定量检测下限,其置信水平约为90%。表3结果表明:本方法中Li、Na、K、Cs在铀氧化物中的方法检测下限分别为:0.015、0.14、0.14、39 μg/g,其中Li、Na、K的检测下限均低于国标分析方法中的检测下限2 μg/g[3],完全能满足分析要求。

表3 方法定量检测下限
2.7 方法准确性
由于目前暂没有相应的标准物质,实验分别采用标准加入法、与国家标准分析方法比对两种方式来验证方法的准确性。
2.7.1加标回收 按照实验方法,选择本底较低的二氧化铀粉末样品,进行Li、Na、K、Cs元素的加标回收实验,结果列于表4。由表4结果可以看出,所有元素的加标回收率均在90%~110%之间,相对标准偏差优于15%(n=6)。方法的精密度高,准确可靠。

表4 加标回收实验结果
2.7.2方法比对 分别采用GB/T 13370-92[3]和本方法进行方法比对。鉴于目前没有以铀为基体的碱金属元素国家标准物质,本工作制备了八氧化三铀为基体的工作标准物质作为比对样品,依据GB/T 13370-92中规定的元素测量范围为2~20 μg/g,制备两个含量水平的工作标准物质。制备流程为将一定量的单元素标准贮备液加入同一生产批次的不含待测元素的二氧化铀粉末样品溶液中,经蒸干、煅烧、均匀性检验制备出两个水平含量的八氧化三铀粉末工作标准样品。分别采用GB/T 13370-92原子吸收分光光度法(AAS)和本方法进行样品处理及分析测定,结果列于表5。由表5可知,在95%置信水平下,经过t检验表明两种方法的测量结果一致。

表5 不同分析方法的测量结果
3 测量结果不确定度的评定[9-11]
根据实验方法的描述和计算公式,影响ICP-AES法测量铀氧化物中碱金属元素的不确定度来源主要有:(1)样品称量的不确定度u(m);(2)体积的不确定度u(V);(3)萃取分离部分的不确定度u(F);(4)测量重复性的不确定度u(ρ)。以下以Li元素测量结果的不确定度评定为例。
3.1 样品称量的不确定度分量u(m)
用天平称取样品的不确定度主要有3个分量:重复性、可读性u1(m)(数字分辨率)、检定或校准u2(m)。重复性不确定度分量在总的重复性不确定度中考虑。一般情况称量进行2次,1次为空盘,1次为毛重,所以上述分量必须计算2次,得样品称量不确定度u(m)=0.13 mg。
3.2 体积的不确定度u(V)
定容体积有3个不确定度分量:校准、温度和重复性。计算校准不确定度时假设为三角分布,计算温度不确定度时假设温度变化是矩形分布,重复性以对1 mL移液管(V1)、100 mL容量瓶(V100)充满10次并称量,计算标准偏差做标准不确定度,得体积不确定度u(V1)=0.005 7 mL、u(V100)=0.074 mL。
3.3 萃取分离铀部分的不确定度分量u(F)
以表4中加标量为1.00 mg/L Li元素的测定结果,计算标准偏差s=0.068 mg/L、回收率平均值为106%,当测量次数为6次时,计算铀与待测元素分离的不确定度分量如下:
(1)
3.4 样品溶液中待测元素测量结果的不确定度分量u(ρ)
样品溶液中Li的测量结果的不确定度分量u(ρ(Li))包括:标准溶液的不确定度u(ρref(Li))与标准曲线的不确定度u1(ρ(Li))两部分。
(1)标准溶液的不确定度u(ρref(Li))包括:标准溶液的不确定度u(ρbiao(Li))、移取标准溶液体积的不确定度和标准溶液定容体积的不确定度。各部分的标准不确定度和相对标准不确定度计算结果列于表6。

表6 标准溶液的不确定度分量
则合成标准溶液的相对不确定度得:
(2)
(2)标准曲线的不确定度u1(ρ(Li))
u1(ρ(Li))即为使用最小二乘法拟合标准曲线时产生的不确定度。校准标准溶液系列分别被测量3次,结果列于表7。

表7 标准曲线的测量结果
标准曲线的线性方程表达式为:
I=aρ+b
(3)
式中:I表示信号强度;ρ表示Li的浓度;a表示斜率;b表示截距。依据最小二乘法分别求得:
经线性拟合得直线方程:I=1 852 726ρ-55 767.5,该线性拟合的不确定度表示为:
(4)

(5)
重复测量样品6次,测量数据列于表8。

表8 Li元素重复测量结果
据表7、8数据可求得:
SR=50 569.591;u1(ρ(Li))=0.021 5 mg/L
(3)合成样品溶液中Li的测量结果的不确定度分量u(ρ(Li))
u(ρ(Li))由式(6)可求得:
(6)
各分量的标准不确定度和相对标准不确定度计算结果列于表9。

表9 输入量的标准不确定度和相对标准不确定度
则Li质量分数w(Li)=21.2 μg/g时,其相对标准不确定度为:
(7)
当置信水平为95%,包含因子K=2时,相对扩展不确定度(Urel(Li))为:
Urel(Li)=0.036×2=0.072=7.2%
4 结 论
采用电感耦合等离子体原子发射光谱法测定铀氧化物中Li、Na、K、Cs元素含量,方法准确可靠,与原子吸收光谱法相比,提高了检测效率,可用于燃料元件制造生产检验工作。
