2型长QT综合征研究进展
2020-10-27郑泽群廉姜芳
郑泽群 廉姜芳
1 概述
长QT综合征(LQTS)主要分为17个亚型,是由15个常染色体上显性遗传相关基因突变导致的,2型长QT综合征(LQT2)是国内最常见的临床亚型,占全部病例的30%~45%[1],由定位于7q35~36上的KCNH2基因(又称hERG)发生突变所致。hERG编码心肌细胞膜上快速延迟整流钾电流(IKr)通道的α亚基,IKr是心肌复极化的主要离子流,突变的hERG基因使hERG通道蛋白功能表现为单倍型不足或显性负效应,导致IKr电流减少或完全消失,心电图表现为QT间期延长和T波异常[2-3]。在休息或睡眠状态下,突然的噪音、惊吓等相关刺激导致患者体内交感系统激活,引发异常钙瞬变,是LQT2发生致命性心律失常如尖端扭转型室性心动过速(TdP)的诱发因素,可突发晕厥甚至心源性猝死。
应用β受体拮抗剂及植入心律转复除颤器(ICD)等治疗对预防高风险患者发生致命性心律失常有一定作用[4]。然而,接受治疗的患者(尤其是女性)仍存在较高生命风险及严重并发症风险[5]。由于病理机制复杂及临床表现多样,LQT2的治疗仍面临严峻挑战。近年来,基因编辑及干细胞技术的发展促进了LQT2相关研究,特别是人诱导多功能干细胞(hiPSC),为测试新治疗方法及疾病建模提供了良好的工具[6]。目前,针对hERG突变相关机制,利用hiPSC等技术的独特优势,不仅能够在体外近似地表达疾病的相关表型[7-8],还可以将纠正LQT2表型的新药转化到临床,并显示了良好的临床适应性[4]。
2 hERG通道
IKr通道由主要的α亚基和辅助性β亚基构成,α亚基由hERG编码,是发挥通道作用的主要功能单位,hERG蛋白含1 159个氨基酸,由6个跨膜片段以及N端Per-Arnt-Sim(PAS)域和C端环状核苷酸结合域(cNBD)组成,4个hERG编码的亚基组成四聚体与辅助亚基共同发挥作用[2,9]。hERG基因经核转录、核糖体翻译后,需经内质网(ER)加工进行核心糖基化、折叠及组装,正确折叠的蛋白质将进入高尔基体进一步复合糖基化,合格的蛋白质会被运输到胞膜发挥作用,不合格的蛋白质将会被送回ER。如果正确折叠仍无法完成,则将滞留在ER中,进而引起内质网应激(ERS),导致蛋白进入泛素蛋白酶体系统(UPS)进行降解[10-12](见图1)。上述hERG蛋白合成及转运过程的各个步骤都需要热休克蛋白(HSP)70/90、钙联蛋白、钙网蛋白、GTP结合蛋白Rab11B、高尔基体基质蛋白GM130等分子伴侣参与[13],众多因素如温度、低氧、pH值、钾浓度都可以影响hERG蛋白成熟[2]。研究显示体内雌激素水平与QT间期有关,雌二醇能够通过增强的雌二醇α受体介导的HSP90相互作用增加hERG通道膜转运及复极化电流[14]。人类J蛋白DNAJB12和DNAJB14通过独立于HSP70的机制促进了Ego相关基因(ERG)K+通道亚基的四聚体组装,过表达DNAJB14可以通过稳定突变的蛋白质改变hERG通道功能缺陷,这证实伴侣蛋白对于亚基稳定性和K+通道的膜定位是必需的[15]。
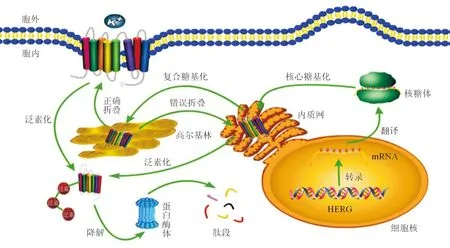
图 1 hERG蛋白的表达调控
IKr电流的减少程度与hERG基因突变部位相关。据报道,52%的hERG基因突变发生在蛋白胞内区域,30%发生在跨膜区域,12%发生在孔区,6%发生在胞外。其中突变发生在跨膜区及孔区(离子进出)导致的IKr电流减少最严重[1],该区域也是众多引发获得性长QT的药物的作用位点[16-17]。功能研究表明,约90%的错义突变通过影响蛋白转运抑制通道功能,这种突变称为Ⅱ类突变;Ⅰ类突变影响蛋白的合成与翻译,而Ⅲ类和Ⅳ类突变则分别通过干扰hERG通道门控特性及减少K+跨膜转运抑制IKr电流[13,18]。
针对不同类型的突变,增加IKr通道电流的干预策略包括通道激活剂、竞争性通道结合、促进hERG蛋白转运、膜定位以及基因编辑等。hERG蛋白转运缺陷是最主要的发病机制[19],对这种突变机制的相关研究已经取得一定成果。
3 LQT2的治疗
LQT2一般采用β受体阻滞剂、交感神经切除等,必要时行ICD植入等治疗,对于预防恶性心律失常发生起到一定作用。目前,在比较成功地揭示了相关致病基因导致不同程度IKr减少的前提下,结合基因编辑及hiPSC技术,为靶向精准治疗提供了实践可能[20]。
3.1 hERG通道变构调节及再转运
hERG通道的变构调节剂能够与其不同位点结合而改变IKr通道活性,从而起到改变其功能表达的作用。LUF7346作为一种新型hERG变构调节剂,被Sala等[21]证实在不同的实验模型中能够逆转先天性和获得性LQTS的表型,在阿司咪唑引起的疾病模型中,应用LUF7346解除了药物的钾通道位阻效应,表明在hERG基因的Ⅲ、Ⅳ类突变时进行hERG通道的变构调节似乎是有益的,这也为LQT2的治疗带来新的思路。
对于广谱蛋白酶体抑制剂ALLN,既往的HEK细胞模型表明该药物能够改变突变的hERG的蛋白表达。Mehta等[22]应用ALLN处理携带hERG A561V错义突变的hiPSC分化的心肌细胞,免疫荧光显示hERG蛋白膜定位增加,膜片钳显示IKr增强,证实了ALLN能够使hERG蛋白再转运,进而纠正LQT2表型。
囊性纤维化跨膜传导调节因子(CFTR)基因突变是造成囊性纤维化的原因,药物鲁玛卡托(VX-809)作为蛋白质折叠分子伴侣,能够促进突变型CFTR(F508del-CFTR)蛋白成熟,起到治疗作用[23-24]。研究显示,纠正F508del-CFTR蛋白转运的小分子药物能够通过不同机制对其他突变蛋白转运缺陷起作用[25]。基于此前提,为了验证鲁玛卡托是否能够纠正hERG蛋白转运,Mehta等[18]成功应用干细胞分化的心肌细胞模型证实鲁马卡托能够纠正LQT2的Ⅱ类突变和表型,促进hERG成熟,使IKr增加,且显示出较强的异常钙瞬变处理能力,在预防心律失常方面比β受体阻滞剂更有效。Schwartz等[4]又进一步应用鲁马卡托+依伐卡托治疗了2例LQT2患者(第1天半量,第2天起全量鲁马卡托800 mg+依法卡托500 mg至第8天),治疗后心电图显示QT间期缩短,且治疗过程中患者仅有腹泻的不良反应。
3.2 规律间隔成簇短回文重复序列(CRISPR)/规律间隔成簇短回文重复序列关联蛋白9(Cas9)、RNA干扰(RNAi)
CRISPR/Cas9技术能够对不同生物体进行基因组编辑,通过选择性敲除及导入基因,在基因复制、转录调控等方面起重要作用[26]。Mesquita等[27]将携带R534C突变的LQT2患者的血单核细胞重编程为hiPSC,并使用CRISPR/Cas9将相同的突变基因插入到对照hiPSC系中,结果表明该技术可以纠正或引入相关基因使hiPSC分化的心肌细胞表型发生改变,进而控制患者的遗传背景和表观遗传变异性。Chai等[28]应用干细胞建立的模型验证了hERG基因同种类型的突变可以导致不同的临床表型,他们还用基因测序鉴别出了新的突变体KCNK17及REM2,并使用CRISPR/Cas9技术将REM2 G96A突变体等位基因校正为野生型,结果动作电位时程和L型钙离子电流(ICaL)恢复并接近正常水平。Garg等[29]利用CRISPR/Cas9编辑疾病特异性hiPSC分化的心肌细胞基因,通过纠正异常突变体及导入相关突变基因到正常细胞系中,实现了突变基因的敲除与导入,并表现为LQT2。这种通过基因编辑改变基因型导致不同的效应,对于先天性LQT2治疗具有重要意义,但基因编辑改造了基因型,基因编辑是否会引起其他基因的异常突变等问题仍需进一步考虑。
结合hiPSC,RNAi也被应用在LQT2的研究中,RNAi能够使同源mRNA高效特异性降解,沉默相关基因表达。Matsa等[30]用突变特异性小干扰RNA(siRNA)处理LQT2患者来源的hiPSC分化的心肌细胞,电生理分析显示IKr电流有所恢复,证实RNAi在体外模型中可以发挥基因调控作用,进而纠正LQT2表型。
包括LQT2在内的多种离子通道病仍有待进一步研究,应用新的研究技术有望推进LQT2的治疗。然而,在精准医疗的背景下,研究成果的临床转化也只有在更加成熟地应用上述技术后,才能使疾病处在更加可防、可控的状态下。
