阿尔茨海默病的内质网应激机制与运动干预研究进展
2020-10-13夏杰,赵娜,王璟,徐波*
夏 杰 ,赵 娜 ,王 璟 ,徐 波 *
阿尔茨海默病(Alzheimer’s disease,AD)作为一种神经退行性疾病,临床上以进行性记忆缺陷和认知障碍为主要表现,病理上以异常磷酸化Tau蛋白(p-Tau)和β淀粉样蛋白(Amyloid-β,Aβ)聚集,以及突触损伤为主要特征。由于AD发病机制不明,目前尚无治疗AD的特效药物,且大多数针对AD病理的药物开发因不能有效改善行为症状而终止于临床3期实验(Gauthier et al.,2016)。因此,探索能缓解AD病理且改善行为表现的药物或非药物干预手段是当前需解决的关键科学问题。
内质网应激(endoplasmic reticulum stress,ERS)是指细胞受到某些因素(如能量匮乏、Ca2+浓度失调、自噬缺陷等)刺激时,内质网稳态改变,错误折叠和未折叠的蛋白质积聚在内质网中,超出了内质网所能承受的蛋白质折叠负荷,诱发细胞做出相应应答,以应对内质网压力的一种状态。其中,未折叠蛋白反应(unfolded protein response,UPR)是ERS状态下恢复内质网稳态的细胞应答类型之一(Walter et al.,2011)。若细胞遭受长期或病理性的ERS,UPR则可过度激活并诱导细胞凋亡(Wang et al.,2016)。研究证实,AD患者和动物模型脑内,Aβ或p-Tau的聚集可导致ERS,引起UPR的过度激活(Gerakis et al.,2018;Hetz et al.,2017);过度激活的UPR信号可通过诱导p-Tau和Aβ生成、抑制突触相关蛋白质合成和扰乱自噬等途径加重AD病理进程(Cai et al.,2016;Guo et al.,2018;Kim et al.,2017;Ma et al.,2013)。提示,ERS在AD病理进程中发挥重要作用。
运动科学领域研究发现,体育锻炼可以延缓老年人认知功能和记忆力下降(Tolppanen et al.,2015)。运动可以缓解AD动物模型的病理表征和行为表现(Hüttenrauch et al.,2016;Moore et al.,2016)。近年已有研究显示,运动可以改善AD脑组织的ERS(George et al.,2018;Kang et al.,2013;Xia et al.,2019)。然而,运动调控脑内ERS在抗AD中的作用机制尚不明晰。基于此,全面分析ERS与Tau病理、Aβ、突触可塑性、细胞自噬和运动之间的关系,提出运动通过调节ERS改善AD的可能机制。
1 ERS与UPR信号转导
ERS导致内质网腔内未折叠或错误折叠蛋白超量积累,引起UPR(Walter et al.,2011)。UPR由内质网的3种跨膜蛋白调控:蛋白激酶样内质网激酶(protein kinaselike endoplasmic reticulum kinase,PERK)、肌醇依赖酶 1α(inositol requiring protein 1α,IRE1α)和激活转录因子 6(activating transcription factor,ATF6)。正常生理状态下,这3种蛋白分别与内质网伴侣分子免疫球蛋白结合蛋白/葡萄糖调节蛋白78(immunoglobulin binding protein or glu‐cose regulating protein 78,Bip/GRP78)相结合,UPR信号抑制。ERS状态下,PERK、IRE1和ATF6分别与Bip解离并激活,启动3条UPR信号。
PERK激活诱导真核翻译起始因子2α(eukaryotic translation initiation factor,eIF2α)的丝氨酸51号位点磷酸化,磷酸化的 eIF2α(p-eIF2α)不能完成 GTP-GDP的交换作用,通过减缓或暂停全局蛋白质的合成(global protein synthesis),降低蛋白质折叠负荷。p-eIF2α可以选择性上调激活转录因子4(activating transcription factor,ATF4)的翻译,ATF4通过促进抗氧化反应、氨基酸生物合成和转运相关基因的转录,缓解 ERS(di Prisco et al.,2014)。ATF4还可促进C/EBP环磷酸腺苷反应元件结合转录因子同源蛋白(C/EBP homologous protein,CHOP)的转录,上调UPR、自噬和mRNA翻译中的功能基因表达(Han et al.,2013)。
IRE1α激活,诱发其核糖核酸内切酶活性,选择性地从X盒结合蛋白(X-box binding protein 1,XBP1)mRNA中切割出26个核苷酸片段,使XBP1翻译并产生转录活性。由IRE1α/XBP1调控的基因不仅增强蛋白质折叠和运输,而且促进蛋白质降解途径,从而减少错误折叠蛋白质的聚集。IRE1α的激活还可介导mRNA衰减(mRNA decay),降低内质网上的蛋白质折叠负荷(Hollien et al.,2006)。
ATF6作为内质网跨膜蛋白,当错误折叠蛋白质在内质网中积累,ATF6转位至高尔基体,被S1P和S2P蛋白酶裂解产生具有转录活性的胞质片段。该胞质片段进入细胞核,一方面诱导内质网伴侣分子的表达,提高内质网蛋白折叠能力。另一方面,提高内质网相关蛋白降解(endo‐plasmic reticulum-associated protein degradation,ERAD)途径中的基因表达,促进未折叠或错误折叠蛋白质的降解(Walter et al.,2011)。
2 ERS介导AD病理
2.1 AD脑内ERS水平升高
研究发现,AD患者脑内Bip/GRP78表达升高,磷酸化的PERK(p-PERK)和p-eIF2α水平升高,并且与Aβ沉积密切相关(Hoozemans et al.,2005;Ma et al.,2013;O’Con‐nor et al.,2008)。在p-Tau水平升高的AD神经元中,PERK和IRE1α信号上调(Nijholt et al.,2012;Stutzbach et al.,2013)。此外,AD患者脑内IRE1α磷酸化(p-IRE1α)水平与其Braak病理分期正相关(Duran-Aniotz et al.,2017)。XBP1启动子多态性与AD风险呈正相关,XBP1蛋白在AD患者颞叶皮质表达上调(Liu et al.,2013)。CHOP在AD患者颞叶皮质也出现表达上调(Lee et al.,2010)。以上研究说明,AD患者脑内出现ERS和UPR信号增强现象。在AD动物模型脑内,该现象得到了验证。5XFAD转基因小鼠中,p-eIF2α蛋白水平和XBP1 mRNA水平升高(Devi et al.,2014;Reinhardt et al.,2014)。在4月龄 APP/PS1小鼠模型中,GRP78、p-PERK、p-eIF2和CHOP基因出现表达升高,并且伴随着年龄增长(7月龄、10月龄),以上基因出现进一步的表达升高(Barbero-Camps et al.,2014)。在3XTg-AD模型中检测到GRP78、XBP1和CHOP的表达升高(Hashimoto et al.,2018;Mota et al.,2015)。以上研究表明,AD脑内出现ERS水平的升高,ERS相关分子可能参与AD病理。
2.2 ERS与Tau病理
Tau病理是AD发病的重要机制。在表现出Tau病理的神经元和胶质细胞中出现ERS标志蛋白的表达升高(Nijholt et al.,2012)。在Tau(rTg4510)转基因小鼠脑内,p-Tau通过与内质网膜和内质网降解相关蛋白的相互作用,破坏内质网稳态,引起ERS(Abisambra et al.,2013)。说明p-Tau可诱导ERS。相反,ERS可通过激活UPR诱导p-Tau生成(Kim et al.,2017)。研究发现,PERK基因的单核苷酸多态性相关的遗传变异是诱发散发性Tau病理发展的危险因素(Hoglinger et al.,2011)。AD患者中发现,p-PERK与活化的糖原合成酶激酶3β(glycogen synthase kinase 3β,GSK3β)在大脑神经元中共同表达(Hoozemans et al.,2009)。在Tau转基因小鼠中发现,p-PERK与p-Tau共定位增加(Ho et al.,2012)。激活UPR信号可通过激活GSK3β,诱导p-Tau的生成增加(Nijholt et al.,2013)。此外,XBP1也在Tau病理调节中发挥作用,在Tau过表达的果蝇模型中,降低XBP1表达可以降低p-Tau所致的神经毒性(Loewen et al.,2010)。
ERS与Tau病理之间存在相互调控的特点,p-Tau的聚集可以诱发ERS,而ERS可以通过上调UPR信号诱导p-Tau生成增加。提示,ERS与p-Tau之间可能形成恶性循环,介导AD病理或行为症状发生。
2.3 ERS与Aβ病理
有研究发现,向培养的神经细胞中加入Aβ寡聚体可以诱导ERS水平升高和细胞凋亡增加(Nishitsuji et al.,2009)。进一步研究显示,Aβ可导致Ca2+通过内质网膜上的Ryanodine受体(Ryanodine receptor,RyR)和三磷酸肌醇受体(Inositol1,4,5-trisphosphatereceptor,IP3R)向胞质和线粒体释放,引起ROS水平升高,导致细胞凋亡增加(Costa et al.,2012;Fonseca et al.,2013)。Aβ还可以激活PERK/eIF2α/ATF4信号通路,促进CHOP基因转录,从而激活细胞凋亡(Xu et al.,2019)。说明Aβ可通过诱导ERS引起AD脑内神经细胞过度凋亡。
近年研究表明,UPR信号可以调控APP代谢,诱导Aβ生成增加。UPR信号启动时,PERK/eIF2α信号通过暂停或减缓翻译过程缓解ERS。但磷酸化的eIF2α却能选择性上调某些mRNA的翻译,其中包括ATF4 mRNA和β-位点 APP水解酶 1(β-site APP-cleaving enzyme 1,BACE1)mRNA的翻译。研究发现,ATF4是早老素1(pre‐senilin 1,PS1)基因的转录因子,ATF4的含量升高可以促进PS1基因的转录(Mitsuda et al.,2007)。在长期ERS状态下,ATF4依耐性的PS1基因转录激活,通过提高PS1的表达而促进γ分泌酶的分泌,诱导Aβ生成增加(Ohta et al.,2011)。Vassar等(2008)发现,BACE1 mRNA 的 5’非翻译区(5’-untranslated region,5’UTR)所含的上游开放阅读框(upstream open reading frames,uORFs)在 eIF2α 磷酸化的情况下可以启动其翻译,从而促进Aβ生成酶β分泌酶的表达。在5XFAD模型小鼠中,PERK/eIF2α信号过度激活,而PERK基因敲减后,海马组织BACE1水平降低,Aβ生成减少(Devi et al.,2014)。采用药理学手段抑制PERK依赖性的eIF2α磷酸化后,APP/PS1小鼠海马组织Aβ生成和沉积减少(Guo et al.,2018)。说明UPR信号上调的BACE1和PS1表达可能通过促进β分泌酶和γ分泌酶的分泌进,诱使APP向Aβ生成途径分解。
综上,长期ERS状态下,UPR信号过度激活,表现为Bip表达升高,p-PERK水平升高,引起下游蛋白eIF2α的磷酸化水平升高。p-eIF2α可通过两条途径共同介导AD病理:1)p-eIF2α促进下游靶分子ATF4的表达,ATF4入核启动PS1基因的转录,使γ分泌酶的表达或分泌增加。ATF4还可以促进CHOP基因转录,诱导细胞凋亡;2)p-eIF2α可调控BACE1基因的翻译,使BACE1的蛋白水平升高,从而增强β分泌酶的表达或分泌。APP在β分泌酶和γ分泌酶的剪切下形成Aβ片段。此外,Aβ的产生可以导致内质网Ca2+失衡,诱导细胞过度凋亡(图1)。

图1 ERS与Aβ病理关系Figure 1.The Relationships between ERS andAβ Pathology
2.4 ERS与突触可塑性
突触可塑性是指突触在神经元持续活动影响下发生的特异性数目、结构和功能的变化,是学习记忆形成的基础。皮质、海马区的突触完整性受损、可塑性异常、密度下降被认为是AD认知障碍的病理机制(Frere et al.,2018)。细胞水平的突触可塑性表现为长时程增强(longterm potentiation,LTP)和长时程抑制(long-term depres‐sion,LTD)。分子水平的突触可塑性体现在新的蛋白质合成(De novoprotein synthesis)和特异性基因表达的调控(Hafner et al.,2019)。
PERK/eIF2α是内质网UPR中通过抑制全局蛋白质合成而缓解ERS的调节信号。然而,长期PERK信号激活可以抑制突触相关蛋白的表达,进而影响突触可塑性。Ma等(2013)研究发现,APP/PS1小鼠脑内p-eIF2α水平升高,新蛋白合成显著减少,而PERK基因缺失可防止新的蛋白质合成的减少,并提高突触可塑性相关蛋白水平和改善LTP缺陷。Yang等(2016)采用基因和药理学手段抑制PERK,发现APP/PS1小鼠中蛋白质合成依赖的代谢型谷氨酸能受体依赖性LTD(metabotropic glutamate receptordependent LTD,mGluR-LTD)损伤得到缓解。在朊病毒小鼠模型中,eIF2α的持续磷酸化显著降低突触蛋白SNAP-25、VAMP-2、PSD-95和 NMDAR1的表达,而降低 p-eIF2α水平可恢复蛋白质翻译率,改善突触缺陷和神经元丢失(Moreno et al.,2012)。说明PERK/eIF2α信号介导的蛋白质翻译抑制可能是AD突触可塑性失调的分子机制。
PERK/eIF2α信号还能通过上调ATF4 mRNA翻译,抑制cAMP反应元件结合蛋白(cyclic-AMP response binding protein,CREB)依赖的突触可塑性。CREB是维持突触功能的关键分子,可以促进突触可塑性相关基因(如BDNF)的表达。研究发现,ATF4是CREB的抑制因子,药理学和基因手段促进eIF2α磷酸化可以提高ATF4的翻译并抑制CREB的表达,从而抑制LTP和长期记忆形成(Costa-Mat‐tioli et al.,2007)。在5XFAD小鼠中,PERK基因单倍剂量不足可以抑制ATF4的表达并促进CREB的表达,改善AD小鼠记忆功能缺陷(Devi et al.,2014)。有研究指出,CREB和PSD-95可以作为PERK的磷酸化底物,PERK的过度激活可以抑制CREB的活性和PSD-95的表达,从而导致突触棘丢失(Sen et al.,2017)。说明PERK信号激活所抑制的CREB转录途径可能是突触可塑性、学习和记忆过程的负调控机制。
研究发现,IRE1α/XBP1信号也在调节突触可塑性中发挥作用。神经元特异性敲除XBP1基因的小鼠记忆形成和LTP受损,而神经元XBP1过表达可上调BDNF的表达,逆转XBP1基因敲除所致的记忆缺陷(Martinez et al.,2016)。Saito等(2018)研究发现,兴奋性突触激活依赖于XBP1的上调,XBP1促进BDNF的转录激活,而BDNF又通过IRE1/XBP1途径以蛋白激酶A(protein kinase A,PKA)依赖的方式驱动其自身的表达。Cisse等(2017)研究发现,海马组织XBP1基因过表达可改善AD小鼠树突棘密度、LTP和空间记忆的缺陷,其机制与XBP1调控Kalerin-7的表达有关。Ma等(2008)指出,Kalerin-7可通过细胞骨架重塑调节树突棘的形成。在AD患者大脑中Kalerin-7蛋白水平降低,而Kalerin-7基因缺失可降低小鼠的突触可塑性并引起记忆障碍(Cisse et al.,2017;Xie et al.,2011)。说明XBP1在突触可塑性和记忆功能中发挥作用,其功能紊乱可能通过影响突触可塑性相关蛋白BDNF和Kalerin-7的表达,介导AD病理。ERS状态下,调节性IRE1依赖的衰变(regulated IRE1-dependent decay,RIDD)可通过介导mRNA衰减,影响mRNA稳定性,进而抑制突触可塑性(Lee et al.,2015;Maurel et al.,2014)。
综上,ERS状态下,PERK/eIF2α信号激活,GTP-GDP交换受阻,降低新蛋白质合成从而抑制突触可塑性。PERK/eIF2α信号激活选择性上调ATF4的表达,通过抑制CREB,影响突触可塑性。IRE1α作为一种核酸内切酶,在ERS状态下激发其活性,一方面通过诱导mRNA衰变,抑制突触可塑性;另一方面,通过调节XBP1的转录活性,参与突触可塑性相关蛋白的表达调控(图2)。
2.5 ERS与细胞自噬
细胞自噬是细胞内的一种分解代谢途径,可将胞质中异常聚集的蛋白质、受损细胞器及其他细胞成分转运至溶酶体进行降解,以维持蛋白质稳态和细胞代谢平衡。生理状态下,内质网通过泛素-蛋白酶体系统(ubiquitin proteasome system,UPS)降解错误折叠的蛋白质,在ERS状态下,UPR激活诱导自噬途径激活,二者相互协调响应并清除错误折叠蛋白质。研究发现,自噬是神经细胞UPR激活后的主要降解途径,然而,在AD脑内表现为过度激活的UPR和无效的细胞自噬,说明ERS-细胞自噬可能在AD病理调节中发生作用(Nijholt et al.,2011;Scheper et al.,2011)。
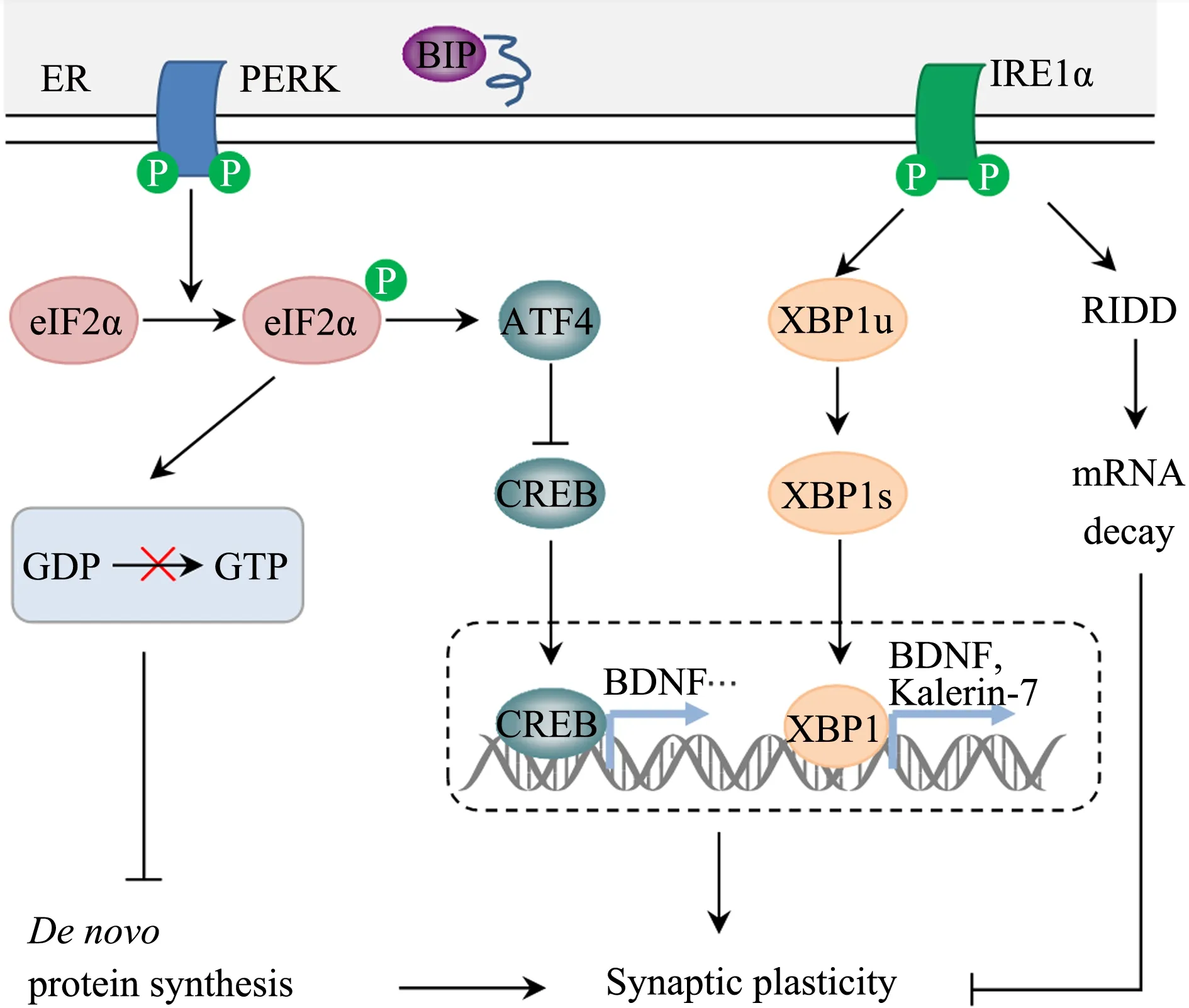
图2 ERS与突触可塑性关系Figure 2.The Relationships between ERS and Synaptic Plasticity
有研究表明,AD脑内自噬异常主要表现为神经元中自噬体大量聚集,自噬体标志蛋白LC3II表达增加,自噬体成核因子Beclin1表达降低,自噬底物标志蛋白P62表达增高,溶酶体活性下降,说明AD脑内自噬障碍,自噬通量降低(Bordi et al.,2016;Lee et al.,2010;Pickford et al.,2008;Sanchez-Varo et al.,2012)。有研究认为,AD脑内细胞自噬障碍不仅与自噬-溶酶体降解途径受损有关,也可能与ERS紊乱有关(Cai et al.,2016;Scheper et al.,2011)。颗粒空泡变性(granulovacuolar degeneration,GVD)被认为是AD脑内自噬过程受损的病理标志,AD患者海马组织GVD区域LC3免疫活性增加,ERS标志物p-PERK免疫活性增加,且LC3与p-PERK共定位增加,说明在AD海马组织中UPR激活与自噬病理学之间存在联系(Nijholt et al.,2011)。
研究发现,在6月龄APP/PS1小鼠皮质区,伴随着UPR信号激活出现PI3K/AKT/mTOR信号增强,p70s6k、ULK1磷酸化和p62水平升高,LC3裂解抑制,说明自噬过程受损;而采用药理学手段抑制PI3K/AKT/mTOR信号后,自噬过程增强,UPR信号下调,说明促进细胞自噬在一定程度上有助于缓解ERS(Guo et al.,2018)。还有研究表明,12月龄APP/PS1小鼠脑内ERS过度激活与自噬相关蛋白Beclin-1、ATG3、ATG7和LC3的表达下调有关;8周的胰高血糖素样肽1(Glucagonlike peptide 1,GLP-1)/葡萄糖依赖性促胰岛素多肽(Glucose-dependent insulino‐tropic polypeptide,GIP)拮抗剂DA-CH3处理可下调ERS并逆转APP/PS1小鼠脑内自噬相关蛋白的表达抑制,进而通过减少Aβ负荷、缓解小胶质细胞激活和提高突触可塑性相关蛋白的表达改善AD小鼠空间学习记忆(Panagaki et al.,2018)。提示,靶向调节ERS和细胞自噬途径可能是缓解Aβ病理和突触功能失调的有效途径。
3 运动通过调节ERS预防和延缓AD的研究进展
3.1 运动通过调节ERS,抑制p-Tau、Aβ生成及其神经毒性
研究发现,长期中等强度的有氧跑台运动可抑制AD小鼠海马组织Tau蛋白的过度磷酸化,改善认知缺陷(Leem et al.,2009;Liu et al.,2013)。长期中、高强度的运动训练均能有效减少脑内Aβ水平,改善AD小鼠的认知功能障碍(Cho et al.,2015;Moore et al.,2016)。Li等(2019)研究发现,高强度间歇运动和中等强度有氧运动均能减少APP/PS1小鼠海马组织Aβ水平,且两种运动方式在减少Aβ水平上没有显著性差异。说明运动可通过减少p-Tau和Aβ改善AD行为症状。
由于p-Tau和Aβ聚集可诱发AD脑内ERS,而过度激活的ERS可通过启动UPR信号诱导p-Tau和Aβ生成增加。因此调节AD脑内的ERS可能为防治AD提供可能。研究发现,AD模型大鼠(Aβ25-35侧脑室注射造模)海马ERS激活,神经细胞凋亡增加;6周的游泳运动可以下调AD模型大鼠海马GRP78的表达,抑制含半胱氨酸的天冬氨酸蛋白水解酶12(cysteinyl aspartate specific proteinase,Caspase12)活化,提高B淋巴细胞瘤2(B-cell lymphoma 2,Bcl2)的表达,同时降低Bcl2-associated X蛋白质(Bcl2-as‐sociated X protein,Bax)的表达,进而抑制海马神经细胞凋亡(刘涛,2012)。在PS2突变小鼠中,Aβ的产生和聚集诱导ERS激活,并由此导致神经细胞过度凋亡和炎症反应,12周的自主跑轮运动可抑制PERK/eIF2α、IREIα/XBP1和ATF6信号,下调JNK-p38MAPK炎症信号和CHOP凋亡信号(Kang et al.,2013)。以上研究表明,ERS参与Aβ诱导的神经毒性,而自主跑轮运动可以缓解ERS介导的神经细胞凋亡和炎症反应。有研究发现,6月龄APP/PS1转基因小鼠海马组织GRP78表达升高,PERK/eIF2α/BACE1和ATF4/PS1信号增强;3个月的有氧跑台运动可以抑制PERK/eIF2α/BACE1和ATF4/PS1信号,并减少海马组织Aβ沉积和可溶性Aβ生成,说明跑台运动降低Aβ负荷涉及调控UPR信号(Xia et al.,2019)。因此,采用有氧运动干预方式,其形式无论是被动的游泳运动、跑台运动,还是主动的跑轮运动,均能有效缓解AD脑内过度激活的ERS和Aβ病理。然而,现有研究中缺乏不同强度(高、中、低)和不同形式(如有氧、抗阻、高强度间歇运动)的运动干预对AD脑内ERS及其相关的Aβ病理表征影响的探索。在未来研究中,对运动干预模式作进一步细化,将能更充分的阐明ERS在运动缓解Aβ病理中的作用特点。
综上,运动缓解Aβ神经毒性和抑制Aβ生成机制中涉及调控ERS,但Aβ生成及其神经毒性与ERS的上下游调控关系尚待深入探讨。另外,运动能否通过调节ERS而减少异常磷酸化Tau,目前尚无直接证据,但运动可以抑制PERK信号,而有证据表明抑制PERK信号可以减少p-Tau生成(Radford et al.,2015)。因此,推测运动也可能通过调节ERS,减少异常磷酸化Tau蛋白。
3.2 运动通过调节ERS,提高突触可塑性
研究发现,4周的中等强度有氧运动可以改善AD大鼠海马齿状回突触传递损伤和LTP抑制,其分子机制涉及纠正突触可塑性相关蛋白CaMKII、Calcineurin和BDNF的表达紊乱(Dao et al.,2016)。6个月的自主跑轮运动可以抑制3XTg AD小鼠海马组织突触素(Synaptophysin,SYN)和PSD-95的表达下调,促进神经生长因子GDNF的表达(Revilla et al.,2014)。提示,运动可以抑制AD脑内突触结构功能蛋白质表达异常。
Cai等(2016)研究发现,运动提高突触可塑性与ERS有关。肥胖大鼠海马组织突触可塑性蛋白表达下调,8周的有氧跑台运动可以显著降低肥胖大鼠海马组织GRP78、p-PERK、p-IRE1α和p-eIF2α的水平,提高突触相关蛋白BDNF和SYN的水平,提示运动可通过调节海马组织ERS而发挥突触保护作用。有研究发现,空间记忆缺陷型小鼠海马组织出现ERS激活和凋亡增加,而运动可以逆转这一过程,其分子机制可能是有氧运动降低海马组织GRP78和CHOP的表达,抑制了caspase-12的活化,并启动CREB/BDNF通路,进而改善小鼠记忆缺陷(Kang,2015)。Lourenco等(2019)研究发现,有氧运动通过诱导Irisin分泌,改善AD模型小鼠突触可塑性和记忆缺陷。在该项研究中,研究人员检测了Irisin对海马神经元eIF2α磷酸化、ATF4表达和新蛋白质合成的影响,发现Irisin可以阻止Aβ寡聚体诱导的p-eIF2α和ATF4表达升高,以及新蛋白质合成的下调(Lourenco et al.,2019)。提示,Irisin对ERS相关分子和突触可塑性的调节作用可能是运动改善AD的解释机制。还有研究发现,12周的有氧跑台运动通过调节海马组织ERS改善长期酒精摄入所致的认知功能缺陷,其分子机制表现为下调GRP78和ATF6,并逆转ERS介导的神经元损伤(George et al.,2018)。
因此,长期中等强度的有氧运动可通过调节ERS提高AD突触可塑性。值得注意的是,有研究发现,急性力竭性运动可通过诱导海马组织ERS的过度激活,从而导致细胞凋亡和突触损伤(Ding et al.,2014)。提示,不同的运动形式可能对AD脑内ERS的干预作用不同。鉴于ERS具有促进细胞存活和诱导细胞凋亡的双向调节作用,及其对不同形式运动作出不同响应等特点,未来研究从不同运动干预方式的角度,探究ERS介导运动调节突触可塑性机制,将具有重要意义。
3.3 运动可调节AD脑细胞自噬与ERS
AD神经元内出现过度激活的ERS和无效的细胞自噬。然而,运动可以调节AD脑内的ERS和细胞自噬。12周的有氧跑台运动可激活NSE/htau23转基因小鼠皮质区mTOR信号,启动自噬相关基因的表达,通过调节细胞自噬清除异常磷酸化Tau蛋白(Kang et al.,2015)。Zhao等(2018)研究发现,12周的有氧跑台运动可以减少APP/PS1小鼠海马组织Aβ沉积、上调LC3II、下调P62和Lamp1水平,说明有氧跑台运动可以通过提高自噬活性,促进Aβ清除。有研究指出,12周的有氧跑台运动还可以抑制APP/PS1小鼠海马ERS和过度激活的PERK/eIF2α信号(Xia et al.,2019)。值得一提的是,上述研究中采用的动物模型和运动干预方案与Zhao等(2018)的研究一致,提示在同等实验条件下,运动可以调节APP/PS1小鼠海马ERS和细胞自噬。机制上,细胞自噬受ERS调控,ERS可通过激活PERK/eIF2α通路诱导自噬启动,eIF2α磷酸化可诱导ATG12依赖的LC3I向LC3II的转化(Kouroku et al.,2007)。PERK/eIF2α的下游分子ATF4和CHOP可介导ERS条件下的自噬基因转录(如 Beclin1、LC3、ATG5、ATG7和ATG12)(B'Chir et al.,2013)。在 AD神经元中发现,PERK/eIF2α信号增强,自噬启动增加,自噬体出现堆积,说明ERS激活可能引起自噬通量的进一步降低(Scheper et al.,2011)。有氧跑台运动可以缓解APP/PS1小鼠海马的ERS、下调PERK/eIF2α信号和提高自噬活性(Xia et al.,2019;Zhao et al.,2018)。说明运动可能通过调节ERS和细胞自噬缓解AD。但目前ERS与细胞自噬是否在运动改善AD病理中相互调控,尚待后续进一步研究。
3.4 运动诱导的能量代谢适应有利于促进AD脑内质网稳态
内质网是控制蛋白质折叠的重要细胞器,内质网会消耗大量的能量以维持腔内蛋白质折叠和修饰活动所需的最佳钙离子浓度和氧化还原电位,良好的能量供应是保证内质网正确合成蛋白的必要条件(van Anken et al.,2005)。然而,在AD脑内出现能量代谢失调、错误折叠蛋白质聚集和突触蛋白合成异常等病理表征。因此,改善脑组织能量代谢可能有利于缓解以上病理。
研究发现,20周的有氧跑台运动通过增加复合物I、复合物IV和ATP合成酶活性,改善线粒体呼吸功能,提高AD小鼠脑组织能量代谢并减少Aβ负荷(Bo et al.,2015)。中等强度有氧运动和高强度间歇运动减少APP/PS1小鼠海马Aβ水平与改善线粒体功能有关(Li et al.,2019)。有氧运动也可以通过激活能量代谢因子AMPK,减少AD小鼠海马Aβ沉积(闫清伟 等,2019)。Yan等(2019)研究发现,12周的有氧跑台运动可以改善APP/PS1小鼠海马线粒体融合分裂,提高ATP水平。结合Coskuner等(2014)的研究,ATP通过作用于Aβ肽链的酪氨酸/丝氨酸位点,降低Aβ蛋白质的错误折叠,继而降低其神经毒性;ATP还可以促进Aβ22-35发生结构上的改变,进而抑制更高分子量的Aβ形成(Exley,1997)。提示,运动改善能量代谢可能有助于减少Aβ的生成和聚集。此外,运动减少异常磷酸化Tau蛋白也与能量代谢的改善有关。36周的有氧跑台运动可上调健康成年大鼠海马组织能量代谢,激活sirt1/AMPK信号通路,并下调p-Tau水平(Bayod et al.,2011)。在STZ诱导的AD模型中,4周的有氧跑台运动可以缓解线粒体功能障碍,提高线粒体内细胞色素C氧化酶活性和ATP合成,并抑制海马组织Tau异常磷酸化(Lu et al.,2017)。说明运动诱导的脑组织能量代谢适应可能是预防和缓解AD脑内错误折叠蛋白质聚集的调节机制。
局部蛋白质合成是神经元突触前和突触后区室普遍存在的特征(Hafner et al.,2019)。运动可以调节突触结构功能蛋白质合成,提高突触可塑性,其机制也与脑组织能量代谢适应有关。一项基于蛋白质组学分析的研究发现,通过质谱分析鉴定出的80个相对丰度较高的蛋白质点中,有约90%的蛋白质与能量代谢和突触可塑性有关(Ding et al.,2006)。其中,运动可以正向调控葡萄糖代谢、ATP合成和能量转导中的关键蛋白,同时运动还上调特异性突触可塑性相关蛋白,如细胞骨架蛋白α-丝联蛋白(α-internexin)和分子伴侣蛋白热休克蛋白8(Heat shock protein 8,HSP8)、热休克蛋白 60Kd蛋白 1(Heat shock 60Kd protein 1,HSP60)和神经元蛋白22(Neuronal protein 22,NP22)。Choi等(2018)研究发现,运动可以增加5XFAD小鼠突触相关蛋白BDNF的水平,并且运动的抗AD效应可以被AMPK激动剂AICAR模拟。提示,运动诱导的脑能量代谢适应可能有助于提高突触可塑性过程。
有研究对3月龄APP/PS1小鼠大脑皮质线粒体相关内质网膜(mitochondria associated endoplasmic reticulum membrane,MAM)进行提取分析,发现MAM中蛋白质合成和折叠相关蛋白、线粒体蛋白质转运和ATP生成相关蛋白出现表达变化(Völgyi et al.,2018)。提示,偶联蛋白质折叠与线粒体能量代谢相关的蛋白表达改变可能是AD的早期事件。然而,在Tg4-42 AD小鼠模型中发现,运动可以上调参与内质网蛋白质合成的伴侣分子(如Cry‐ab、Hspa1b、Hsp90ab1、Dnajb1、Dnajb2、Hsph1、Pdia3、Pdia4、Pdia6、Sec61g和Herpud1)的表达(Hüttenrauch et al.,2016)。这些伴侣分子在蛋白质合成过程中起着重要的作用,有助于蛋白质的正确易位、修饰和折叠,其中一些分子伴侣,如Hsp90ab1,已被证实在AD中表达抑制(Brehme et al.,2014)。说明运动可以改善AD脑内质网蛋白质折叠和质量控制能力。
因此,运动对脑组织能量代谢的调控可能有助于改善AD脑内质网蛋白质折叠和质量控制能力,从而减少AD脑内错误折叠蛋白质和增加突触可塑性。未来探讨运动脑组织能量代谢适应与内质网稳态之间的关系,将为揭示运动减少AD脑内错误折叠蛋白质聚集和提高突触可塑性相关蛋白质合成机制提供新命题。
4 小结与展望
综上所述,得到运动、ERS与AD病理相互关系(图3):AD病理主要表现为Tau蛋白过度磷酸化、β淀粉样蛋白沉积和突触损伤。p-Tau和Aβ聚集可以诱导ERS产生,而长期的ERS可通过PERK和XBP1信号诱导p-Tau生成增加;通过PERK/eIF2α信号上调BACE1和PS1的表达,诱导Aβ生成;通过eIF2α磷酸化抑制全局蛋白质合成、ATF4信号抑制CREB依赖转录途径和IRE1α/XBP1信号调节BDNF和kalerin-7的表达,影响突触可塑性。运动可通过抑制ERS,减少p-Tau和Aβ聚集,提高突触可塑性和自噬活性等途径缓解AD,其机制可能是运动促进能量代谢适应,改善蛋白质折叠和质量监控的内质网环境,减轻ERS,从而防止错误折叠蛋白质的产生和突触相关蛋白表达紊乱。
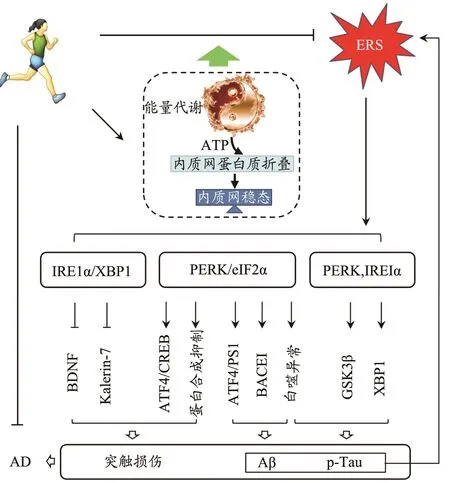
图3 运动、ERS与AD病理关系Figure 3.The Relationships between Exercise,ERS andAD Pathology
未来研究有以下问题需进一步明确:1)ERS是AD病理的促成机制,还是AD病理条件下的适应性反应?2)能量代谢失衡是否是AD脑内错误蛋白聚集的深层机制?3)运动调控ERS的分子机制是什么?不同形式的运动干预对ERS介导的AD病理影响是否有所不同?结合ERS的生物学特点,未来研究对运动干预作进一步细化,是否能为“精准运动医疗”提供科学依据?4)运动改善脑组织能量代谢是否有助于缓解ERS和AD相关的蛋白质稳态失衡?这些问题的探讨将有助于阐明运动防治AD的本质效应,而基于能量代谢与内质网稳态控制关系的探索,将有助于发现新的AD药物作用靶点。
