三萜类化合物抗病毒的构效关系及其作用机制研究进展
2020-09-29范炳芝王一鑫廉霄甜谢维松于洋梁建华
范炳芝,王一鑫,廉霄甜,谢维松,于洋,梁建华
(北京理工大学化学与化工学院,北京100081)
引 言

图1 四环三萜和五环三萜的代表性骨架Fig.1 Representative skeletons of tetracyclic triterpenes and pentacyclic triterpenes
三萜类(triterpenoids)化合物衍生自植物体内甲羟戊酸(mevalonate pathway, MVA)或甲基赤藓醇−4−磷 酸(2−C−methyl−D−erythritol 4−phosphate,MEP)途径合成的异戊烯基焦磷酸(3−isopentenyl pyrophosphate, IPP)或二甲基烯丙基焦磷酸(dimethylallyl pyrophosphate, DMAPP)[1]。通常由六个五碳的异戊二烯单元组成,以游离形式或与糖结合成苷存在于甘草、柴胡、人参、三七和黄芪等植物中;后者因具有亲水亲脂两性,能产生肥皂般泡沫而得名皂苷。三萜类化合物种类繁多,其骨架超过100 种[2],以四环三萜和五环三萜最为常见。四环三萜的骨架类型主要有羊毛脂甾烷型(lanostane)、达玛烷型(dammarane)、大戟烷型(euphane)、原萜烷型(protostane)、葫芦烷型(cucurbitane);五环三萜的骨架类型主要有齐墩果烷型(oleanane)、乌苏烷型(ursane)、羽 扇 豆 烷 型(lupane)和 木 栓 烷 型(friedelane)(图1)。三萜类化合物具有广泛的药理活性,如抗肿瘤[3−4]、抗病毒[5−6]、抗菌[7−8]、抗炎[9−10]以及免疫调节[11]等,因此备受研究学者们的关注。近年来,多种三萜类化合物及其衍生物被证明具有显著和广谱的抗病毒活性,而有望作为新型广谱抗病毒药物应用于临床治疗。
病毒是引发人类疾病和威胁生命健康的主要原因之一,根据mRNA 生成机制不同可分为DNA 病毒、RNA 病毒和逆转录病毒。其中,人类免疫缺陷病毒(human immunodeficiency virus, HIV)、流感病毒(influenza virus)、乙型肝炎病毒(hepatitis B virus,HBV)、丙型肝炎病毒(hepatitis C virus,HCV)、冠状病毒(coronavirus)、呼吸道合胞病毒(respiratory syncytial virus, RSV)、人 乳 头 瘤 病 毒(human papillomavirus, HPV)和单纯疱疹病毒(herpes simplex virus, HSV)等是容易造成全球流行性疾病的病原体。截止到2016 年,已有90 多种抗病毒药物被批准用于临床,表1 列举了部分常用的抗病毒药物。这些药物通过抑制病毒感染过程中的不同阶段(包括病毒的渗透、脱壳、合成核酸、将DNA 整合进入宿主的基因组、结构蛋白的表达和组装、病毒从宿主中释放以及成熟),以达到抗病毒的效果[12−13]。药渡公司统计2018年全球抗病毒市场规模超过550 亿美元,其中抗HIV 为300 多亿美元,抗肝炎市场为195亿美元。突发高传染性病毒感染引发的大流行疾病可造成较高的死亡率,如由2019新型冠状病毒2019−nCoV(后命名为SARS−CoV−2)引发的肺炎,截止到2020年5月2日,全球确认感染已超过330 万,累计死亡超过23 万例,截止到2020 年8月8 日,全球累计病例已迅速攀升至1946 万,累积死亡超过72万例,且病毒的突变率较高易导致耐药病毒株的出现,需要不断开发出新的抗病毒药物。三萜类化合物不仅能够在病毒复制早期抑制病毒的吸附和侵入宿主细胞,还可以在病毒侵染细胞后抑制其复制或成熟的过程,从而能有效保护宿主细胞免受病毒感染[14]。截至目前,尚未有具有抗病毒作用的三萜类药物上市,只有Bevirimat(编号曾为PA−457、BVM、DSB)、RPR103611、BMS−955176(后来编号为GSK−3532795)、GSK−2838232、MPC−9055(未公布结构)、ME3738(图2)进入到临床试验阶段,除了GSK−2838232 正处于临床IIb 期外,其他的几个均已被终止发展[6]。本文将对三萜类化合物抗病毒作用的构效关系及其作用机制进行简要综述,重点介绍其对艾滋病毒、流感病毒、冠状病毒和乙肝/丙肝病毒的活性和作用机制,为三萜类抗病毒药物的设计和开发提供参考。

表1 部分常用于临床病毒感染治疗的药物Table 1 Some anti-virus drugs commonly used clinically

图2 进入到临床试验阶段的三萜类抗病毒药物Fig.2 Triterpenoid anti−virus drugs entering clinical trials
1 三萜抗HIV 的构效关系与作用机制

图3 甘草酸及其天然衍生物1和2的化学结构Fig.3 Chemical structures of glycyrrhizic acid and its natural derivatives 1 and 2
HIV 是一种具有包膜的逆转录病毒,其病毒颗粒由两个单链RNA 基因组、逆转录酶(RTase)和整合酶组成。其包膜包含gp120 和gp41 两种糖蛋白(gp120 和gp41 由前体蛋白gp160 被蛋白酶切割而得),gp41 是跨膜蛋白,gp120 位于表面,它们在病毒传播过程中与宿主CD4+受体结合而进入宿主细胞;包膜内侧是蛋白p17 形成的球形基质以及蛋白p24 形成的锥形衣壳,病毒的基因组和酶处于衣壳内部[15]。HIV 主要通过血液和体液传播,可分为HIV−1 和HIV−2 两种亚型,HIV−2 的致病性要比HIV−1 低得多,并且其分布范围受到限制[16]。疫苗的研发受制于HIV 病毒的变异以及病毒潜伏期等问题,以至于目前仍然没有有效的HIV 疫苗[17−18]。抗HIV药物的分子靶标涉及HIV生命周期中的许多关键步骤,从病毒吸附宿主细胞开始,细胞蛋白、CD4+和趋化因子受体是HIV 进入细胞的关键因素;与受体结合后,病毒通过膜融合进入细胞;随后在细胞中脱壳,并将其RNA 基因组逆转录为DNA;后续还有核易位、染色体整合、RNA 转录、蛋白质合成、病毒装配、病毒后代出芽和成熟。根据药物对HIV 作用部位的不同,可分为:进入阶段抑制剂(阻断HIV 吸附或膜融合)、逆转录酶抑制剂、蛋白酶抑制剂、成熟阶段抑制剂和双功能抑制剂五种类型[19−20]。五环三萜类化合物存在多个位置可用于侧链修饰,如C−3、C−17 和C−28,其修饰结果可使化合物的抗病毒活性和选择性得到大幅度提高。
1.1 齐墩果烷型三萜抗HIV 的构效关系与作用机制
1.1.1 甘草酸及其衍生物 甘草酸(glycyrrhizic acid, GL)(图3)是甘草根提取物中的主要活性成分,是被检测到具有抗HIV−1 活性的首批天然糖苷之一,其抗HIV−1 活性的机理与其聚阴离子的结构有关(甘草酸具有3 个羧基结构),因此可阻断HIV−1 颗粒与细胞CD4+受体结合并干扰蛋白激酶C(protein kinases C, PKC:PKC 为HIV−1 颗粒与细胞CD4+受体结合所必需的酶)的活性来抑制病毒对靶细胞的吸附过程[20−21]。另外,从甘草根中提取分离得到的化合物1 和2(图3)表现出较好的抗HIV 活性,对HIV 的IC50值 分 别 为29.5 μmol/L 和41.7 μmol/L[22]。
此外,一些化学修饰的甘草酸衍生物也是有效的HIV−1 和HIV−2 体外抑制剂,如化合物3 和4(图4)即是由甘草酸衍生而得,两者均对HIV 的蛋白p24 表现出一定程度的抑制作用,其中3 的ID50值(抑制50%p24 活性的浓度)为0.35 μmol/L,4 的ID50值为0.09 μmol/L,但是两者的抗病毒活性均弱于一线药物叠氮胸苷(叠氮胸苷相应的ID50值为0.014 μmol/L)[23]。

图4 甘草酸衍生物3和4的化学结构Fig.4 Chemical structures of glycyrrhizic acid derivatives 3 and 4
1.1.2 齐墩果酸及其衍生物 齐墩果酸(oleanolic acid,OA)(图5)广泛分布于木犀科、龙胆科、葫芦科等植物体内,具有抗病毒[24]、抗菌[25]、抗肿瘤[26]和抗炎[27]等多种生物活性。从人参根部分离出的齐墩果酸衍生物5(图5),是第一个含有聚乙炔结构且能够抑制HIV−1 复制的三萜类化合物,其IC50值为11.1 μmol/L[28]。表明聚乙炔结构可能能够促进化合物对于HIV−1 的抑制作用,而具体机制还需进一步研究。
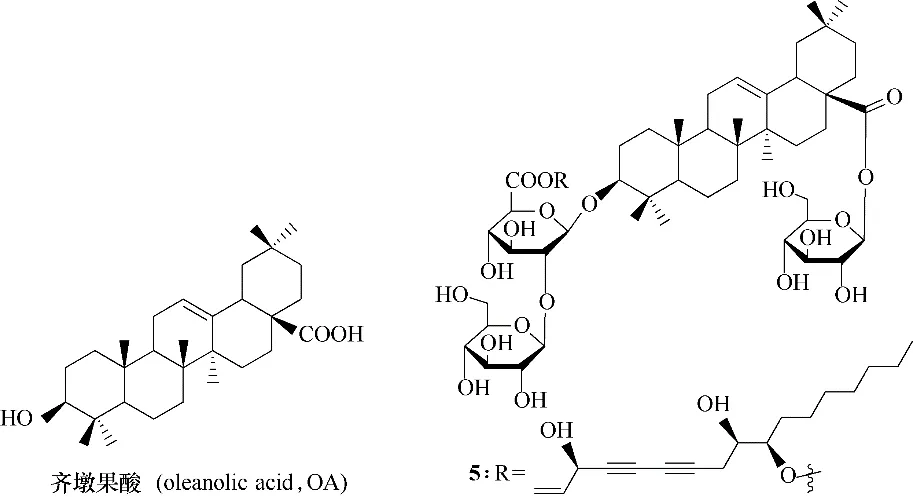
图5 齐墩果酸与其天然衍生物5的化学结构Fig.5 Chemical structures of oleanolic acid and its natural derivative 5
齐墩果酸有三个可供修饰的位点,C−3 位的羟基、C12−C13 位的烯烃和C−28 位的羧基,对不同的位置进行不同的修饰将会不同程度地影响化合物对于HIV−1复制阶段的抑制活性。将C−3位羟基酯化(图6,化合物6~9),会导致化合物活性下降;将C12−C13 位烯烃还原为烷烃得到化合物10(图6),其活性较母体增加3 倍;用适当的酸酐在10 的C−3位进行酯化(图6,化合物11~15),除11 外,其余化合物的活性均有所提升,其中15 的活性最为显著;将齐墩果酸C−28 位的羧基转化为氨基甲基(图6,化合物16),然后再在C−3 位羟基和C−28 位氨基同时引入3′,3′−二甲基戊二酸或3′,3′−四亚甲基戊二酸(图6,化合物17 和18),可使活性增加十倍以上。在6~18 这批化合物中,仅化合物15 的抗病毒活性略优于药物叠氮胸苷,化合物15 的EC50值(抑制50% HIV 复制所需的浓度)为0.0039 μg/ml,叠氮胸苷的EC50值为0.01 μg/ml,但是化合物15 的细胞毒性却远远高于叠氮胸苷[29]。因此,C12−C13 位的构型可能是促使化合物抗HIV 活性提升的关键,另外,对C−3位的羟基和C−28位的羧基进行适当的修饰也可提升化合物的活性。然而,如何能够降低该类化合物的细胞毒性,还有待探索。
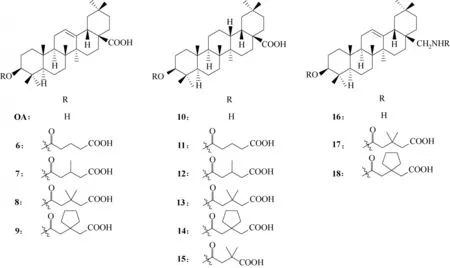
图6 化合物6~18的化学结构Fig.6 Chemical structures of compounds 6—18
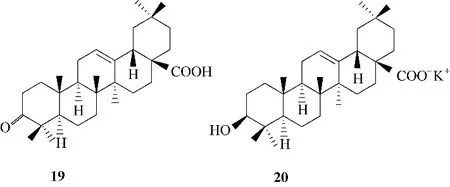
图7 化合物19和20的化学结构Fig.7 Chemical structures of compounds 19 and 20
另外,对于齐墩果酸C−3位修饰的衍生物,具有3′,3′−二甲基琥珀酸取代基的化合物活性优于3′,3′−二甲基戊二酸取代基的化合物[30]。将齐墩果酸C−3 位羟基进行氧化得到3−酮基齐墩果酸(图7,化合物19),依然维持抗HIV 活性,但是其细胞毒性大幅度增强;用氢氧化钾对齐墩果酸进行处理,得到齐墩果酸的钾盐(图7,化合物20),具有明显提升的抗HIV 活性,EC50值(抑制50% HIV 复制所需的浓度)为0.5 μg/ml[31]。19 和20 的抗HIV 活性虽然较母体有所提升,但是仍劣于药物叠氮胸苷(叠氮胸苷相应的EC50值为0.012 μg/ml),并且细胞毒性远高于叠氮胸苷。
1.2 乌苏烷型三萜抗HIV的构效关系与作用机制
提取分离自日本路边青的熊果酸(ursolic acid,UA)和山楂酸(maslinic acid)(图8)显示出有效的抑制HIV−1 蛋白酶的活性[32]。Kashiwada 等[33]基于先前对齐墩果酸衍生物的研究经验[31],制备了C−3 位取代的熊果酸衍生物21~28(图9),研究显示,除23外,其余衍生物的细胞毒性均弱于母体,而抗HIV的活性无明显的提升,此现象与齐墩果酸的C−3 位单侧链修饰衍生物活性研究类似,即对C−3 位进行单侧链修饰对抗HIV 活性无明显改善[29];另外,熊果酸的钾盐(图9,化合物29)的细胞毒性也明显增强。未见将C12−C13 烯烃还原或C−3 与C−28 双侧链取代的熊果酸衍生物,无法得知其修饰是否能够明显提高化合物抗HIV的活性。

图8 熊果酸和山楂酸的化学结构Fig.8 Chemical structures of ursolic acid and maslinic acid
1.3 羽扇豆烷型三萜抗HIV 的构效关系与作用机制
白桦酸(betulinic acid,BA)又名桦木酸(图10),提取自棒花蒲桃的叶子,是最早被确认为具有抗HIV 活性的羽扇豆烷型五环三萜类化合物[34]。白桦酸最初被鉴定为抑制HIV−1 复制的弱抑制剂,而在此基础上开发的白桦酸C−3 位的衍生物Bevirimat(PA−457)(图2)则是一种有效的抗HIV−1 制剂,作用于病毒的成熟阶段[35]。已进入到临床试验II 期,但是由于病毒结构蛋白对其敏感性降低,于2010年终止研究[6]。对白桦酸的结构修饰主要包括三个方面:C−3 位羟基的修饰(成熟阶段抑制剂)、C−28 位羧基的修饰(进入阶段抑制剂)以及C−3 位羟基和C−28 位羧基同时修饰(进入及成熟阶段双功能抑制剂)。
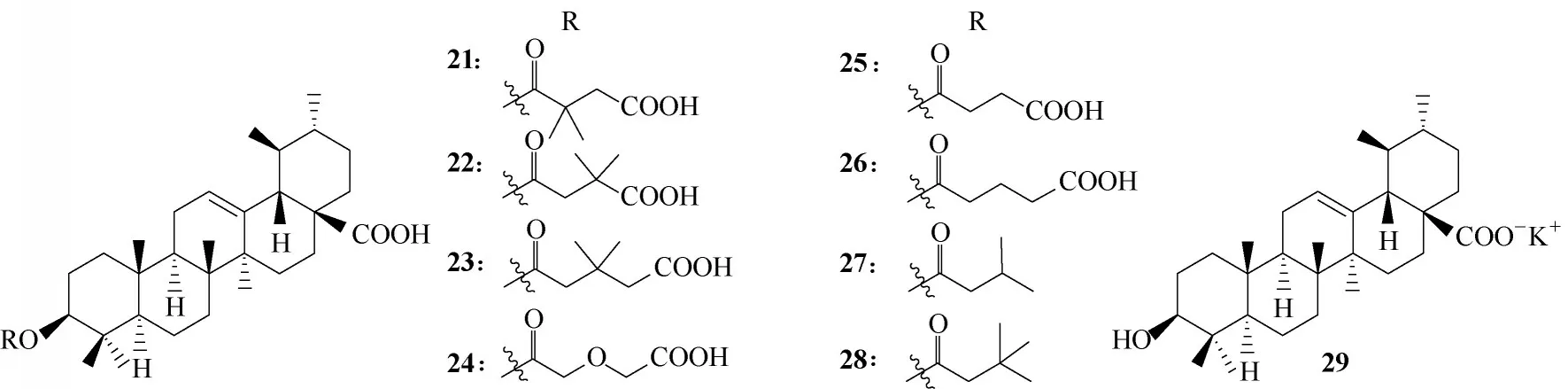
图9 熊果酸衍生物21~29的化学结构Fig.9 Chemical structures of ursolic acid derivatives 21—29
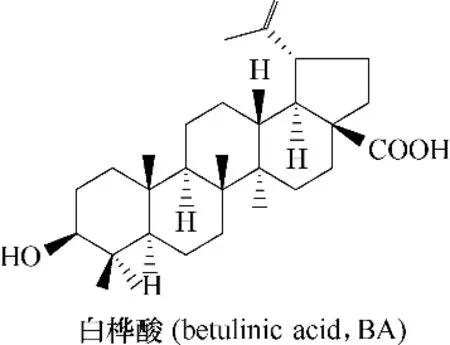
图10 白桦酸的化学结构Fig.10 Chemical structure of betulinic acid
Evers 等[36]发现白桦酸C−3 位羟基对抗HIV 活性的重要性:将构型由3β 转变为3α,差向异构导致活性降低至1/10;将3−羟基氧化为3−酮基,活性无明显变化;而脱水形成2,3−烯烃或3 位引入甲氧基或C−2 位引入羟基都会导致体外活性丧失。然而,Sun 等[37]提出C−3 位酰基侧链对活性的提升至关重要。在白桦酸C−3 位引入3′,3′−二甲基琥珀酸或酰基,可提升其抗HIV−1 活性,其作用机制为干扰了正常的病毒结构蛋白Gag 的加工,从而阻止病毒的成熟过程[38−41]。
在三萜类化合物的C−28 位羧基进行修饰所得的衍生物,已被证明能够有效抑制HIV−1 与宿主细胞膜融合的过程[20]。其中,最初报道的白桦酸衍生物RPR103611(图2)的靶点与HIV−1 跨膜糖蛋白gp41 有关[42−44]。尽管其在体外显示出强大的抗病毒活性,但由于体内药效特性较差,Rhone−Poulenc(现为赛诺菲−安万特)终止了RPR103611 的临床开发[45]。
HIV−1与宿主细胞的融合过程涉及跨膜糖蛋白gp41 的两个螺旋区域:一是N 端七肽重复序列(heptad repeat)HR1(也称为N 螺旋或N 肽),另一个是C 端七肽重复序列HR2(也称为C 螺旋或C 肽)。当病毒与宿主细胞膜融合时,HR1 和HR2 会形成一种具有融合活性的构象,称为“发夹三聚体(trimer−of−hairpins)”。该结构由三个来自gp41 单体肽的6个α−螺旋束组成,这种结构是许多包膜病毒所共有的结构[46−47]。发夹三聚体对于病毒与宿主细胞膜融合过程极其重要,因而成为了抗病毒药物的潜在靶标。Si 等[48]首先以CEM 4 作为宿主细胞,对化合物30 和31(图11)的HIV−1 病毒侵染细胞的抑制活性进行了鉴定,发现化合物30 的抑制活性高达95%,化合物31 的抑制活性高达85%。另外,通过光交联技术证明白桦酸衍生物31 的重氮基团能够与HR2的M626 残基紧密结合,同时,模拟对接结果(图12)显示化合物30 和31 与HR2 之间存在多重疏水相互作用,另外,30 和31 侧链的3−OH 位于HR2 的极性残基S649 和Q650 附近,羧基位于由D624、N625 和T627 组成的极性口袋中,证实了HIV 跨膜糖蛋白gp41中的HR2是三萜类化合物的直接靶标。
对白桦酸的C−3 位羟基和C−28 位羧基同时进行修饰,所得的衍生物32和33(图13)兼具抑制HIV病毒进入和成熟两个功能,作用位点分别为膜表面蛋白gp120 和结构蛋白Gag[49];衍生物34~37(图13)表现出极强的抗HIV−1 活性,其中37 的活性最好,EC50值(抑制50% HIV−1 IIIB 复制的浓度)为0.006 μmol/L,而药物叠氮胸苷的EC50值仅为0.056 μmol/L。表明C−28 位侧链的原子长度对提高抗病毒效力具有重要意义,同时侧链结构含酰胺基团对活性很重要[44,50]。Sun 等[51]在研究白桦酸衍生物IC9564(图14)时,认为C−3位游离的羟基以及C−28位侧链末端游离的羧基是抗HIV活性所必需的,且C−28氨基烷烃侧链上亚甲基的数目对活性至关重要。
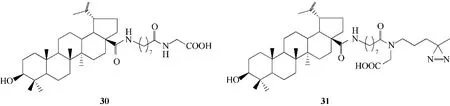
图11 白桦酸衍生物30和31的化学结构Fig.11 Chemical structures of betulinic acid derivatives 30 and 31

图12 化合物30(a)和31(b)与HR2的对接结果[48]Fig.12 Docking results of 30(a)and 31(b)with HR2[48]
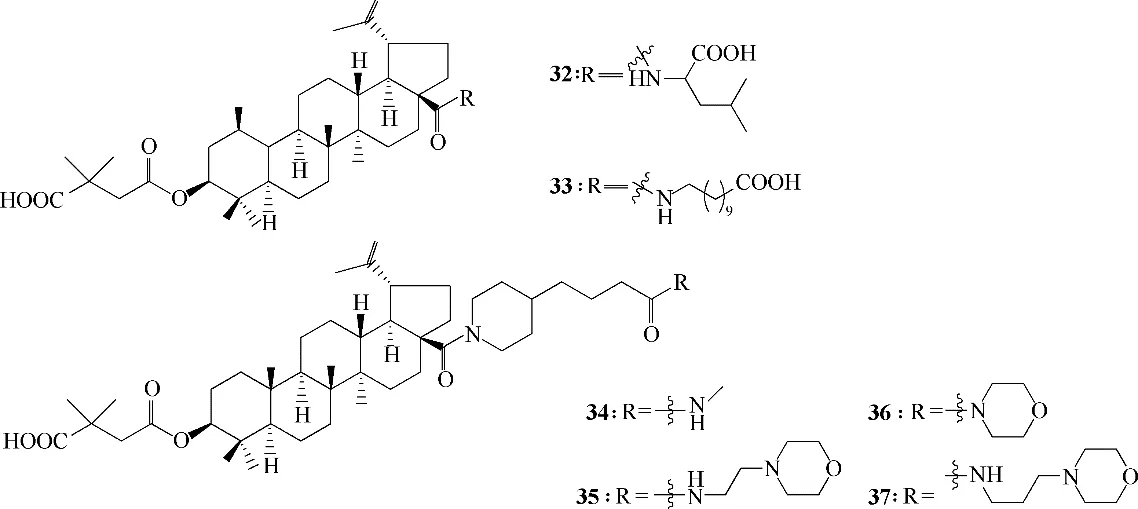
图13 白桦酸C−3,C−28双取代衍生物32~37的化学结构Fig.13 Chemical structures of betulinic acid C−3,C−28 disubstituted derivatives 32—37

图14 白桦酸衍生物IC9564的化学结构Fig.14 Chemical structure of betulinic acid derivative IC9564
Ren等[52]基于琥珀酸侧链的变化,设计的C−3和C−17双取代衍生物BMS−955176(后来编号为GSK−3532795)(图2),是有效的HIV 成熟阶段抑制剂,已进入临床IIb 期试验。BMS−955176 的结构改造历程表明白桦酸C−3 和C−17 位的取代基对于获得具有所需病毒学和药代动力学特征的分子至关重要[53−55]。另外,基于BMS−955176 的分子结构所开发的类似物38~63(图15),其构效关系证实了BMS−955176为抑制HIV−1成熟阶段的最佳化合物[56]。但是由于其严重的副作用,于2016年终止发展。
Qian 等[50]提出白桦酸C−19 位的异丙烯基可能不是活性药效团,但是对该位置进行适当修饰可以改善药物的水溶性,进而改善药物的药代动力学性质;但是在C−30 位引入羟基会导致抗病毒活性降低,表明异丙烯基附近存在氢键供体对于活性具有不利影响,这与文献[36]报道的结果一致。图16 为文献总结的白桦酸衍生物的抗HIV构效关系。
Li等[57]认为对白桦酸等天然产物进行修饰是一条提高抗HIV 效力的可行性路径,但并不是唯一的路径,还可以通过改变母体结构来改变化合物的活性和生物学特性,例如将氟原子引入化合物Bevirimat 结构(图2)中,会影响化合物的亲脂性、构象柔韧性、代谢稳定性等性质。氟具有独特的物理特性,将其掺入分子对药物设计非常重要,并且临床治疗中存在许多含氟的药物[58]。但是在白桦酸衍生物的研究中,将氟引入Bevirimat 的结构中(图17,化合物64~72),化合物的活性均没有得到改善[57]。化合物65 与Bevirimat 的区别仅在于前者在C−2 位引入氟取代,活性消失表明C−2 位的质子可能对抗HIV 活性起关键作用;而化合物68 的抗HIV−1 活性与Bevirimat 相当,表明C−30 位烯丙基的变化对活性无明显的影响,即C−30的烯丙基可能不是抗HIV活性的关键药效团,这与文献[50]报道的研究结果一致。
1.4 其他三萜类化合物抗HIV 的构效关系与作用机制
根据杂合物的设计理念,用化学键连接两种类型的HIV 抑制剂,如HIV−1 逆转录酶抑制剂和HIV−1 蛋白酶抑制剂,两种酶抑制剂的偶联不仅可以降低病毒耐药性的出现速度,而且可以增强抗HIV的活性。部分三萜类衍生物抗HIV活性较低可能是细胞通透性差所致,因此制备了三萜类与叠氮胸苷的偶联物73 ~75(图18),它们的IC100(完全抑制HIV−1 诱导MT−4 细胞病变的最低浓度)分别为0.589 μmol/L、0.370 μmol/L、0.469 μmol/L,表明可开发为抗HIV−1制剂[59]。
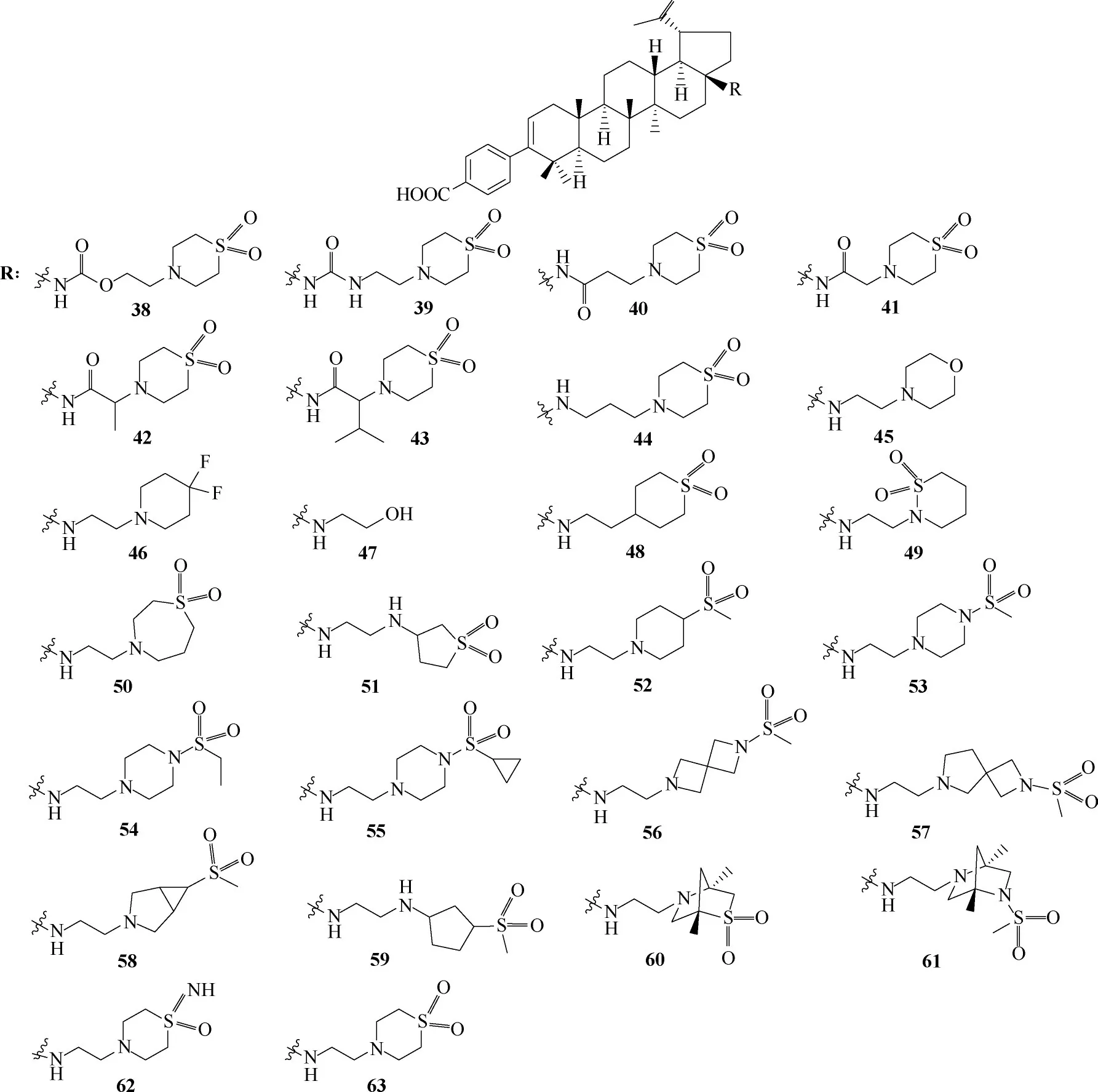
图15 BMS−955176的类似物38~63的化学结构Fig.15 Chemical structures of analogs 38—63 of BMS−955176

图16 白桦酸衍生物抗HIV活性的构效关系[50]Fig.16 Structure−activity relationships of betulinic acid derivatives against HIV[50]
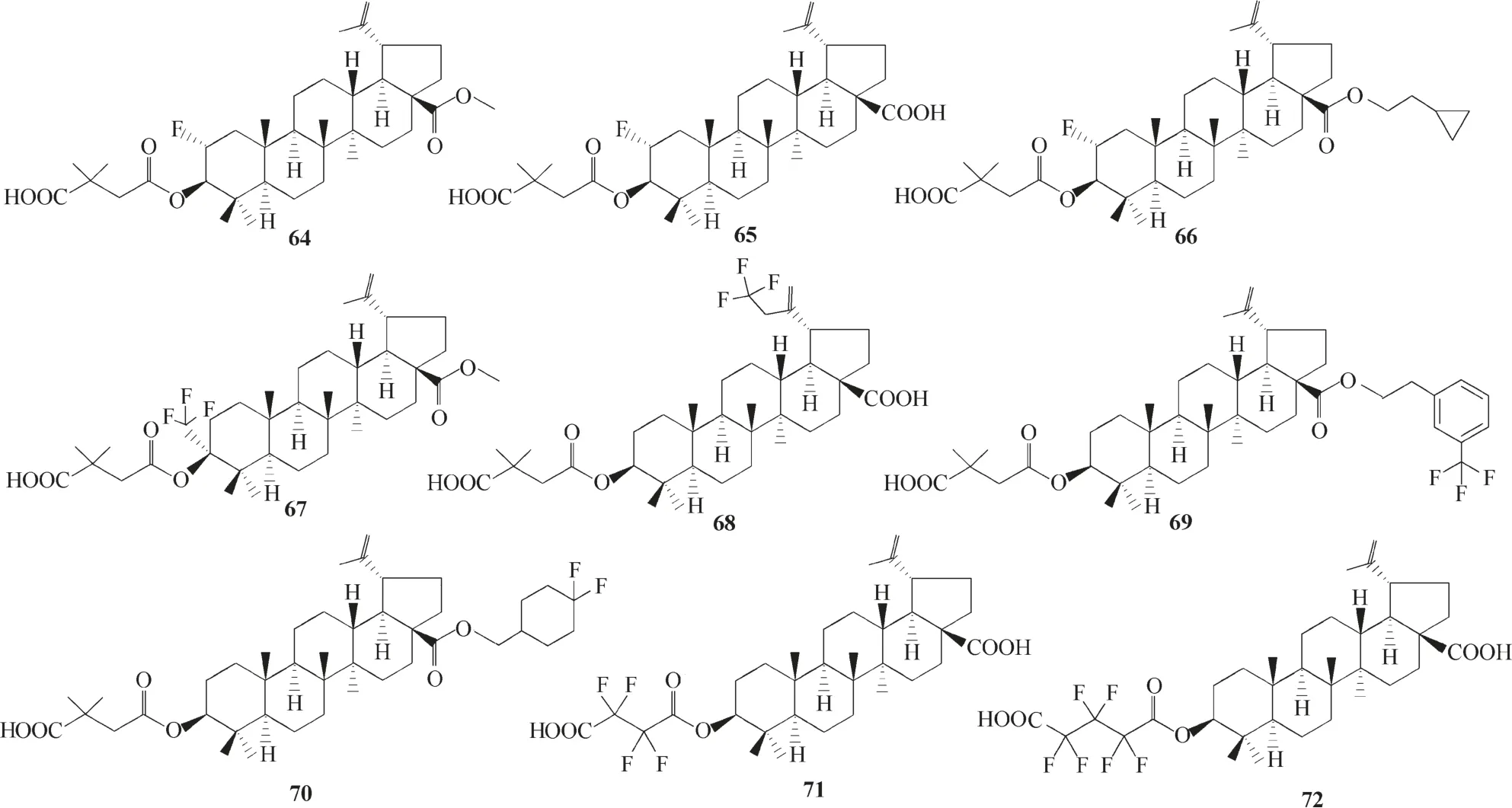
图17 Bevirimat氟化衍生物64 ~72的化学结构Fig.17 Chemical structures of Bevirimat fluorinated derivatives 64—72
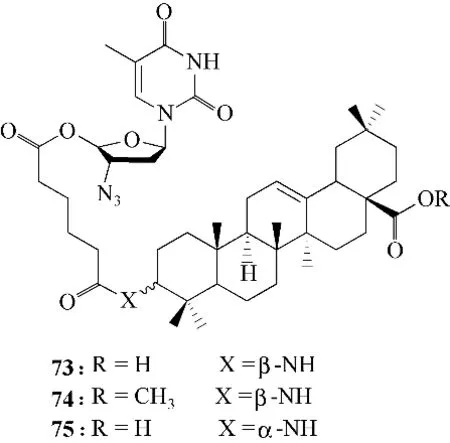
图18 三萜类与叠氮胸苷的偶联物73 ~75的化学结构Fig.18 Chemical structures of conjugates of triterpenes andazidothymidine 73—75
以C−3 位为醛基的2,3−开环三萜(图19,76 和77)为原料,经过格式试剂处理之后氧化得到具有甲基酮结构的化合物78 和79(图19),79 经过碱处理之后得到A 环中具有α,β−烯腈取代的化合物80(图19),实验结果显示78 和80 均具有抑制HIV−1体外繁殖的活性,78 的EC50值(抑制50% HIV−1 诱导MT−4 细胞病变所需的浓度)为7.2 μg/ml,80 的EC50值为39.92 μg/ml[60]。
2 三萜抗流感病毒的构效关系与作用机制
流感病毒(influenza virus)属于正粘病毒科,是一种具有多种抗原特性的RNA 病毒,病毒包膜含血凝素蛋白(HA)和神经氨酸酶(NA,又称唾液酸酶)两种结构蛋白,流感病毒通过其表面的血凝素蛋白,识别和结合呼吸道表面的唾液酸(sialic acid)受体而引发感染[61]。作为常见的病原体,流感病毒可通过飞沫传播,可导致人类和动物严重的急性呼吸道感染疾病。流感病毒感染的第一步是由血凝素介导病毒颗粒附着到宿主细胞上,进而与宿主细胞唾液酸受体结合,导致病毒被内吞。神经氨酸酶抑制剂(例如奥司他韦和扎那米韦)和M2 离子通道抑制剂(例如金刚烷胺和金刚乙胺)是两类临床上用于对抗流感病毒的药物,可阻止病毒进入细胞[62]。M2蛋白是一个离子通道,允许质子穿过,M2离子通道抑制剂则阻断离子通道活性,并防止病毒RNA 释放到宿主细胞质中[63];神经氨酸酶抑制剂靶向神经氨酸酶蛋白,抑制其活性,并导致子代病毒无法从宿主细胞中脱离[64]。目前,已经识别出四种类型的流感病毒,即甲、乙、丙和丁型流感病毒,其中甲型流感跨多物种传播且易突变,分为16 个HA 和9 个NA亚型,危害最大。
2.1 齐墩果烷型三萜抗流感病毒的构效关系与作用机制
2.1.1 甘草酸及其衍生物 甘草酸显示出广泛的抗病毒谱,在对其抑制流感病毒的机制的研究中,发现其可以与细胞膜相互作用,从而阻止病毒被内吞[65]。从甘草根部提取分离得到的化合物81 ~84(图20)对H1N1 流感病毒表现出良好的抑制活性,其IC50值与药物磷酸奥司他韦相当[22]。此外,甘草酸衍生的85 ~88(图21)均表现出有效的抗流感活性,其体外活性较一线药物金刚烷胺高出十倍以上[66],表明甘草酸衍生物具有开发成为抗流感病毒制剂的潜力。
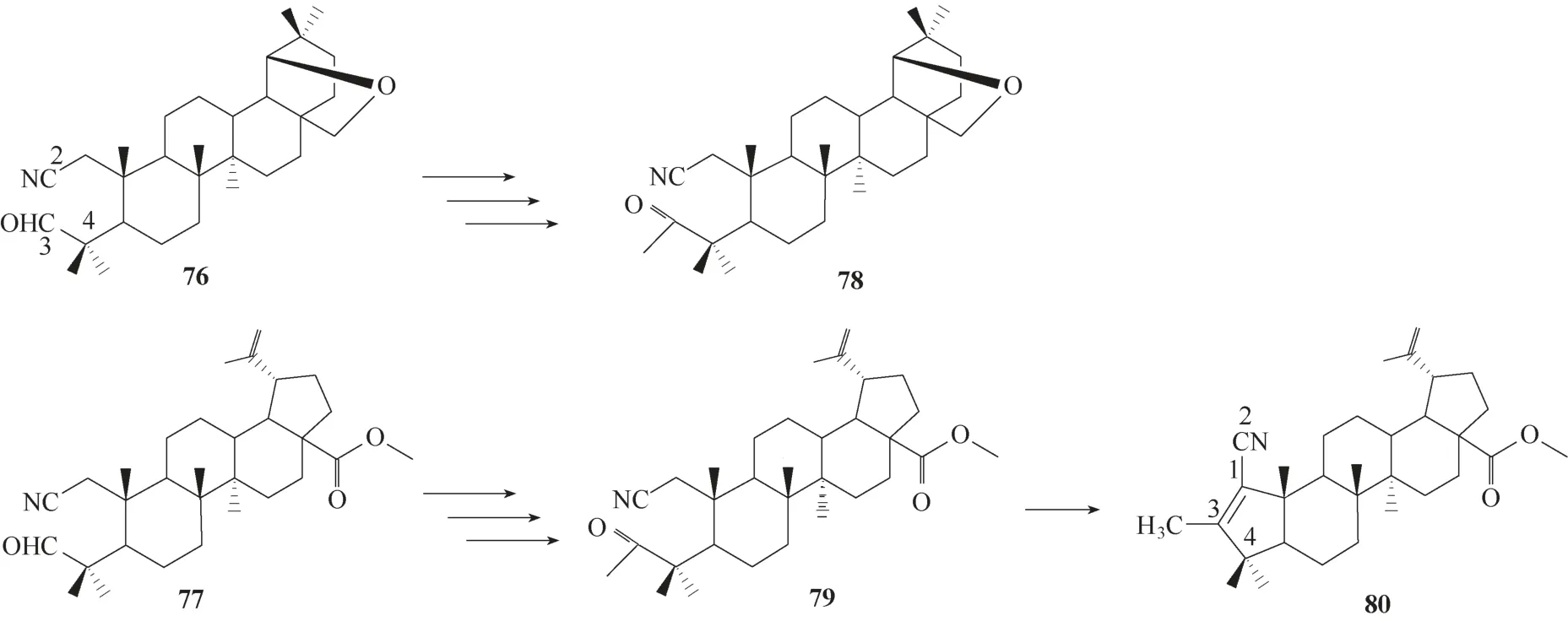
图19 化合物76~80的化学结构Fig.19 Chemical structures of compounds 76—80
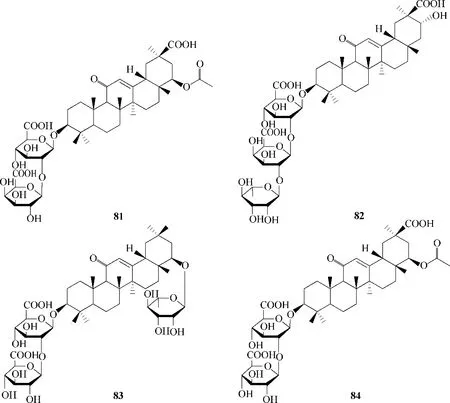
图20 天然产物81 ~84的化学结构Fig.20 Chemical structures of natural products 81—84
唾液酸存在于各种动物组织的糖蛋白和糖脂的糖链末端,因其具有抗病毒等多种生物活性,并且唾液酸−双黄酮类共轭物具有抗病毒活性,而被应用到三萜衍生物的研究中。甘草酸衍生物与唾液酸的缀合物(图22,89和90)和齐墩果酸衍生物与唾液酸的缀合物(图22,91和92)均显示出对流感病毒结构蛋白NA 的抑制活性,并且对流感病毒的增殖有明显的抑制作用[67]。
环糊精(cyclodextrin,CD)是一类高度水溶性和生物相容性的环状寡糖,与三萜类化合物缀合可改善三萜化合物的水溶性,以增强其生物利用度。其中甘草次酸与环糊精的缀合物93 ~98(图23)具有良好的抗病毒活性的同时,均较其母体表现出更高的水溶性和更低的细胞毒性[68]。
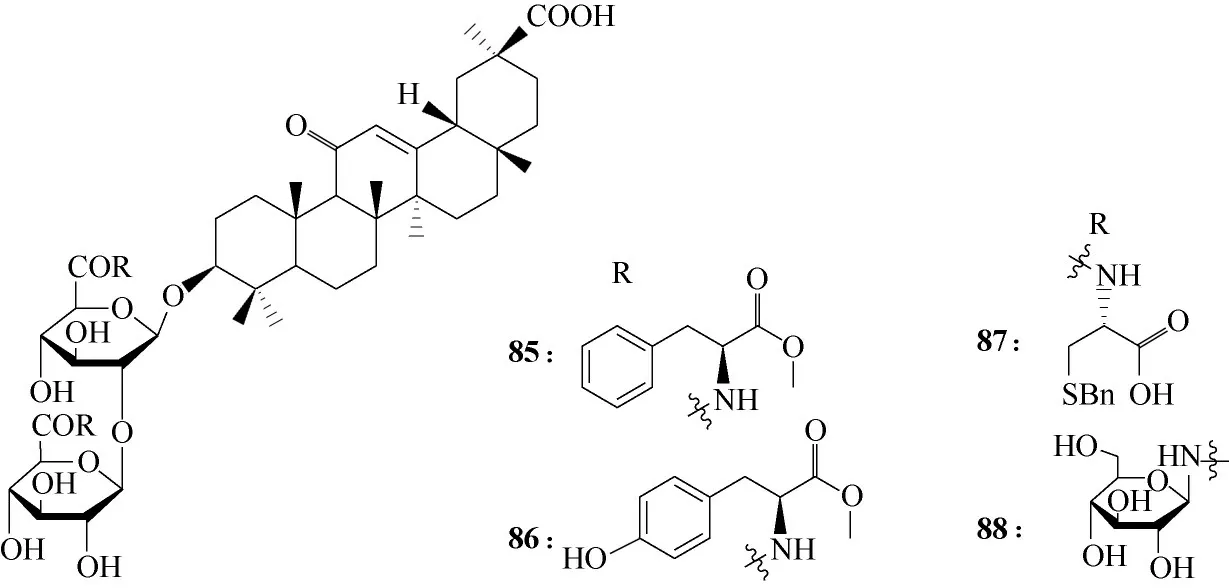
图21 甘草酸衍生物85 ~88的化学结构Fig.21 Chemical structures of glycyrrhizin derivatives 85—88

图22 三萜与唾液酸缀合物89 ~92的化学结构Fig.22 Chemical structures of the conjugates 89—92 of triterpene and sialic acid
2.1.2 齐墩果酸及其衍生物 已知神经氨酸酶抑制剂和M2 离子通道抑制剂分别通过抑制结构蛋白NA 和离子通道达到阻止病毒进入细胞的效果[62]。然而,耐药性流感病毒的出现限制了这些药物的使用。为了寻找新型的抗病毒药物,研究者们对三萜类药物进行抗病毒疗效研究。Yu 等[69]通过分别测定在不同的时间里、在添加化合物99(图24)的条件下,流感病毒核衣壳蛋白在感染细胞中的表达水平,以此初步判断药物的作用阶段。结果表明,化合物99 的抗病毒活性仅在病毒发展周期的早期有效,因此推测该化合物作用于病毒附着或与宿主细胞融合阶段。基于流感病毒传播感染的第一步是结构蛋白HA 与宿主细胞唾液酸受体结合,因此研究者们将化合物的作用靶标锁定为结构蛋白HA,以破坏HA 与唾液酸受体的结合过程,有效阻止流感病毒进入宿主细胞[69−76]。血凝抑制(HI)、表面等离子共振(surface plasmon resonance,SPR)和分子对接等实验结果表明,化合物99(图25)[69]、100(图26)[70]、101(图27 和图28)[71]、102(图29 和图30)[72]、103 ~108(图31 和图32)[73]等化合物能够与病毒包膜结构蛋白HA 紧密结合,破坏HA 与唾液酸受体的相互作用,从而破坏病毒与宿主细胞的结合,达到抗病毒效果,其体外抗病毒效果与一线药物奥司他韦相当。为了评价化合物99 在体内的抗流感病毒作用,Yu 等[69]通过对感染A/WSN/33 流感病毒的小鼠进行给药实验,发现化合物99 对病毒的感染状况有改善作用,且其治疗效果呈现剂量依赖性。此外,与未经药物处理的小鼠相比,化合物99 处理的感染小鼠死亡率明显下降,表明化合物99 可以在体内保护小鼠免受流感病毒的感染。以上研究证明了三萜类化合物可以作为抗流感病毒的新型先导化合物。
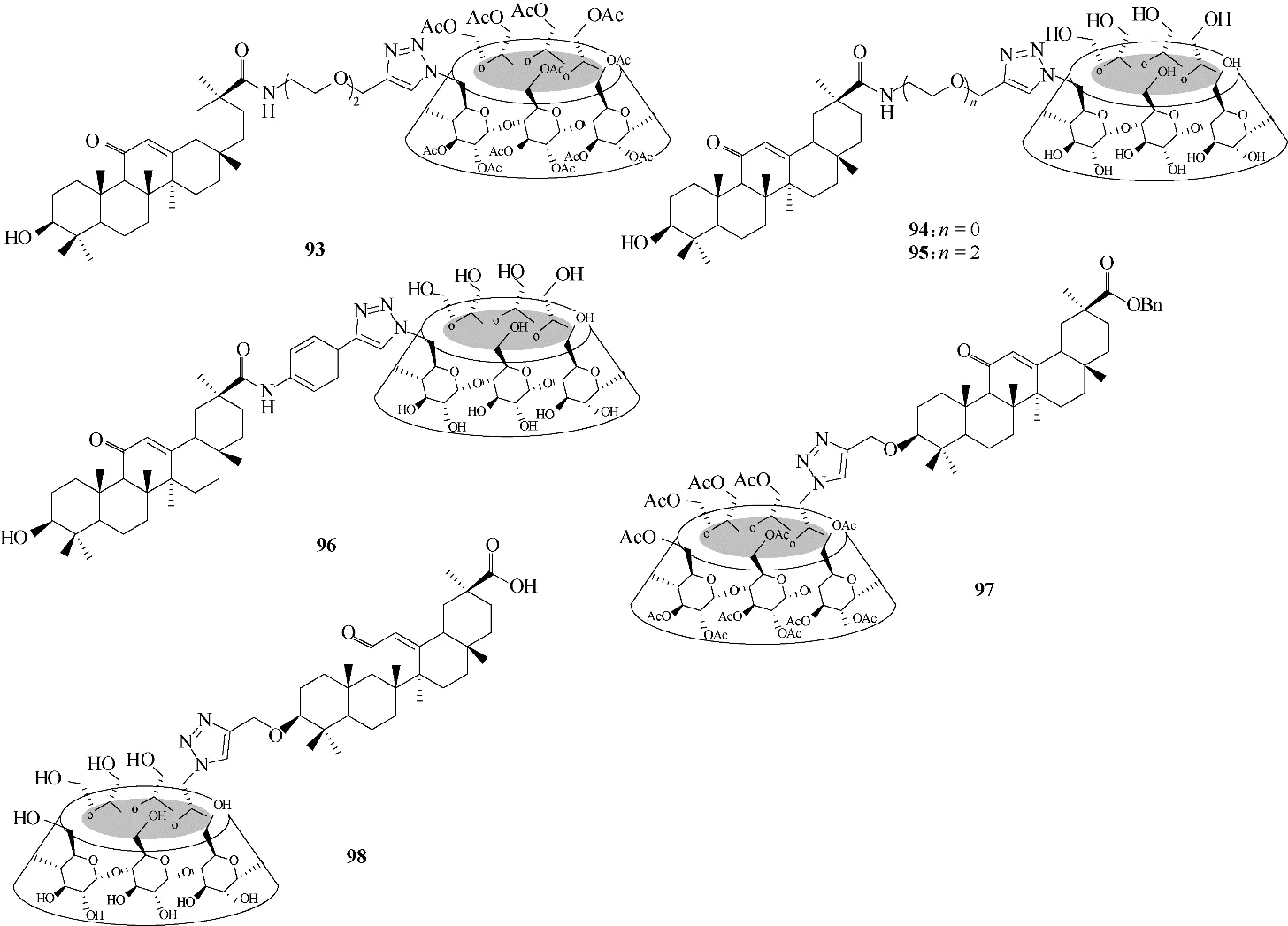
图23 甘草次酸与环糊精的缀合物93 ~98的化学结构Fig.23 Chemical structures of the conjugates 93—98 of glycyrrhetinic acid and cyclodextrin
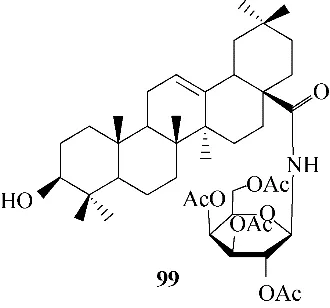
图24 化合物99的化学结构Fig.24 Chemical structure of compound 99
流感病毒与宿主细胞膜的融合过程与HIV 一样,同样涉及蛋白HA 的HR1 和HR2 两个α−螺旋区域。光交联反应结果显示,化合物109(图33)能够与HR2 区域中N471 和I476 之间的残基紧密结合;同时,模拟对接实验结果(图34)显示,109与HR2之间存在多重疏水相互作用,且109 侧链的重氮基团与HR2 的残基N472 之间形成氢键[48]。这两个实验说明HR2 是三萜类化合物抑制流感病毒与宿主膜融合的直接靶标。
研究[77]表明,具有三糖配基的化合物110(图35)对H5N1 流感病毒进入宿主细胞的过程表现出良好的抑制活性,其IC50值为7.2~8.0 μmol/L,进一步的分析研究表明,三糖配基对于110 的活性至关重要[74,78−79]。同样具有三糖配基的化合物111(图35)与HA 的 分 子 对 接 显 示,111 与HA 的Lys26、Asn27、Asn50、Asn53四个氨基酸残基形成比较强的氢键作用(图36),且分子对接结果与111 对HA 的抑制活性实验结果具有一致性,表明该类化合物的作用靶点可能为蛋白HA[74]。
为了进一步探究三糖配基与三萜化合物的构效关系,选取齐墩果酸为母体,固定C−3位为三糖配基,改变三萜骨架或对C−17 进行不同侧链修饰(图37,化合物112 ~124),表明C−28 位的C O 基团对维持活性的重要性(113 vs 120),对该位置羧酸进行酯化或酰胺化修饰有利于提升抗病毒活性,同时降低细胞毒性;另外,异构的3−α构型有利于降低化合物的细胞毒性(113 vs 122),而三萜的结构骨架对抗H5N1 流感病毒活性没有显著影响(123 vs 124),揭示了结构修饰对抗病毒活性的重要性[75]。此外,化合物118 与HA 的模拟对接结果与文献[74]报道化合物111 的结果具有一致性,更加证实了C−3 多糖取代对提升抗病毒活性的重要性。
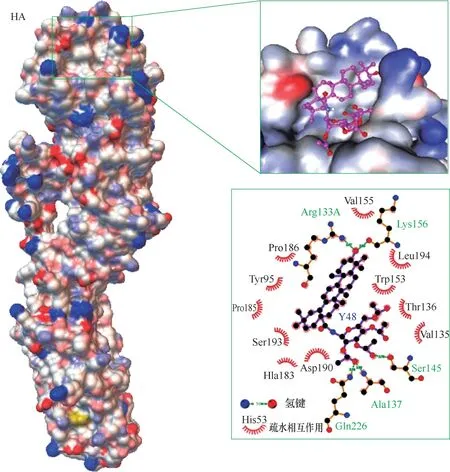
图25 化合物99与HA的对接结果[69](图中显示,99占据唾液酸受体的保守结合口袋,并且99的C−3−羟基与残基Arg133、Lys156形成氢键;99的β−糖基部分的2′−羰基与Ala137、Gln226形成氢键,β−糖基部分的3′−羰基与Ser145形成氢键。另外,在99及其结合口袋之间存在多重疏水相互作用,即与残基Val155、His183、Ser193、Pro185、Tyr95、Pro186、Asp190、Trp53、Thr136、Val135和Leu194之间存在疏水相互作用)(ACS出版社授权使用)Fig.25 Docking results of compound 99 with HA[69](It shows that 99 occupies the conserved pocket of sialic acid receptor,and the 3−OH of 99 forms hydrogen bond with the residues Arg133 and Lys156;the 2′−carbonyl of the β−galactose moiety of 99 forms hydrogen bond with Ala137 and Gln226,and the 3′−carbonyl of the β−galactose moiety forms hydrogen bond with Ser145.In addition,there exist hydrophobic interactions between 99 and residues Val155,His183,Ser193,Pro185,Tyr95,Pro186,Asp190,Trp53,Thr136,Val135 and Leu194)(With the permission of ACS Press)
图26 化合物100的化学结构Fig.26 Chemical structure of compound 100

图27 化合物101的化学结构Fig.27 Chemical structure of compound 101

图28 化合物101与HA的对接结果[71](101的C−3位羟基与Gly225形成氢键,糖基部分的2′−羟基和3′−羟基也分别与Arg133、赖氨酸Lys156和Gly158形成氢键;C−28位的羰基和糖基的6′−羟基与Ser193形成氢键。此外,三氮唑部分与Val155和Leu194之间存在堆积相互作用)Fig.28 Docking results of compound 101 with HA[71](The 3−OH of 101 forms hydrogen bond with the residues Gly225,and the 2′−OH and 3′−OH of the glycosyl moiety also form hydrogen bond with Arg133,Lys156,and Gly158;the carbonyl at C−28 of 101 and 6′−OH of glycosyl moiety form hydrogen bond with Ser193.In addition,there is a stacking interaction between the triazole portion and Val155 and Leu194)
图29 化合物102的化学结构Fig.29 Chemical structure of compound 102
为了了解三萜C−3位多糖配基的类型、构型、链长度以及糖键和化合物抗病毒活性之间的相关性,设计了化合物125~134(图38)[76]。其构效关系表明葡萄糖上C−2′和C−4′羟基上的取代基对抗病毒效力具有重要作用(125 vs 126,127,128,129);取代基处于葡萄糖C−2′或C−4′,对活性也会产生极大的影响(133 vs 134);三萜的结构骨架对抗H5N1 流感病毒活性没有显著影响(130 vs 132),这与先前文献[75]的研究结果一致。
为了改善三萜类化合物的水溶性,设计了环糊精与三萜的缀合物。所有缀合物的水溶性均得到改善,化合物135 ~137(图39)对甲型H1N1 流感病毒有较强的抑制活性,并且与蛋白HA 具有较高的亲和力[80−81]。
2.2 羽扇豆烷型三萜抗流感病毒的构效关系与作用机制
从枣树(Zizyphus jujuba Mill)分离得到的白桦酸已被证明可通过抗血栓形成而抑制流感病毒的感染[82]。从日本桤木(Alnus japonica)树皮中提取分离的成分138(图40)对流感病毒表现出明显的抑制作用,其 对 流 感 病 毒KBNP−0028 的EC50值 为12.5 μmol/L,遗憾的是,细胞毒性较大[83]。从活性角度考虑,该化合物有发展成为抗病毒药物的潜力,但是还需进一步优化以降低其对细胞的毒性作用。
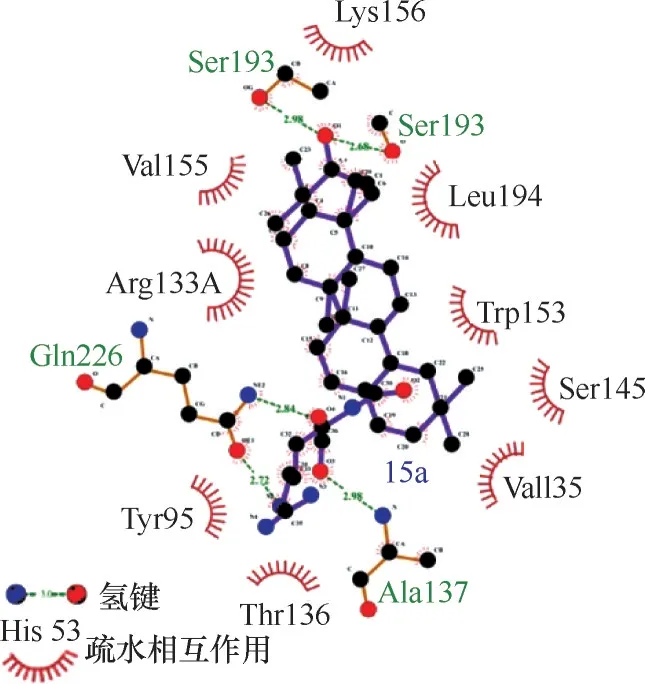
图30 化合物102与HA的对接结果[72](102的3−羟基与Ser193形成氢键,侧链部分与Gln226和Ala137形成氢键。另外,102与结合口袋中的Lys156、Leu194、Trp153、Ser145、Val135、Thr136、Tyr95、Arg133、Val155残基存在疏水相互作用)Fig.30 Docking results of compound 102 with HA[72](The 3−OH of 102 forms hydrogen bond with Ser193,and the sidechain forms hydrogen bond with Gln226 and Ala137.In addition,there exist hydrophobic interactions between 102 and the pocket formed by Lys156,Leu194,Trp153,Ser145,Val135,Thr136,Tyr95,Arg133,and Val155)
由白桦酸衍生而来的化合物139(图41)具有显著的抗流感病毒活性,其EC50值(抑制50%流感病毒诱导MDCK 细胞病变的浓度)较药物磷酸奥司他韦的更低(磷酸奥司他韦相应的EC50值为12.5 μmol/L)。139 与HA 的对接结果(图42)显示,139 的C−3位羟基与Lys222 形成氢键,C−17 位羰基与His180形成氢键,而侧链部分与HA 之间存在氢键相互作用、π−π 堆积相互作用、疏水相互作用,极大地增强了化合物与HA 之间的结合力,这也解释了为什么该化合物的活性较高的内在原因[84]。
3 三萜抗冠状病毒的构效关系与作用机制
冠状病毒是一种具有包膜的正链RNA 病毒,其包膜包含膜糖蛋白(membrane glycoprotein, M)、包膜蛋白(envelope protein, E)和棘突糖蛋白(spike glycoprotein,S)。冠状病毒基因组RNA 可被翻译产生两个复制酶多蛋白,称为pp1a和pp1ab,其中pp1a包含一个木瓜样蛋白酶(papain−like protease,PLpro)和 一 个 小 RNA 病 毒 3C 样 蛋 白 酶(picornavirus 3C−like protease, 3CLpro,也称主要蛋白酶或Mpro),3CL 蛋白酶和PL 蛋白酶对病毒的复制必不可少[85]。
图31 化合物103 ~108的化学结构Fig.31 Chemical structures of compounds 103—108
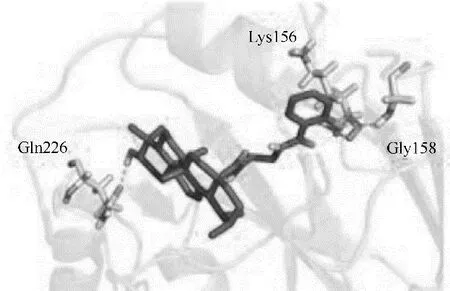
图32 化合物106与HA的对接结果[73](106的3−羟基与Gln226形成氢键,侧链部分与Lys156和Gly158形成氢键)Fig.32 Docking results of compound 106 with HA[73](The 3−OH of 106 forms a hydrogen bond with Gln226,and the sidechain forms hydrogen bond with Lys156 and Gly158)

图33 化合物109的化学结构Fig.33 Chemical structure of compound 109

图34 化合物109与HR2的模拟对接结果[48]Fig.34 Docking results of 109 with HR2[48]
2019新型冠状病毒(2019−nCoV,引发新型冠状病毒肺炎COVID−19)是目前已知的第7种可以感染人的冠状病毒,其余6 种分别是HCoV−229E、HCoV−OC43、HCoV−NL63、HCoV−HKU1、SARS−CoV(引发重症急性呼吸综合征)和MERS−CoV(引发中东呼吸综合征)。2019年12月以来新型冠状病毒肺炎造成全球健康威胁,到目前为止,只有较少的治疗方法和药物被开发出来,如干扰素和甘草酸。甘草酸是首批在体外发现对SARS−CoV 有活性的化合物之一,Cinatl等[86]首先报道了甘草酸及其衍生物具有抗SARS−CoV 活性,其不仅可以抑制病毒复制过程,还能在病毒生命周期的开始阶段抑制病毒的吸附和渗透。曹敏杰等[87]采用RT−PCR 和Western blot 技术,证明了甘草酸可有效抑制猪呼吸道冠状病毒(PRCV)的复制过程。

图35 化合物110和111的化学结构Fig.35 Chemical structures of compounds 110 and 111
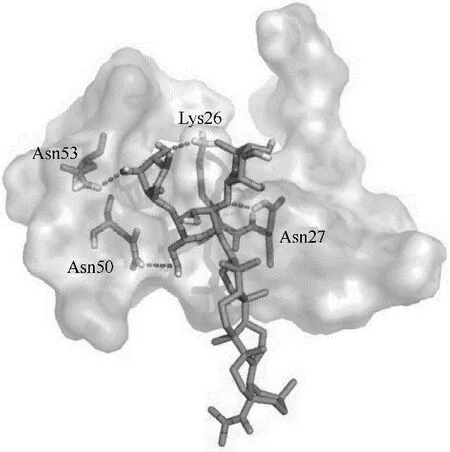
图36 化合物111与HA的分子对接结果[74]Fig.36 Docking results of compound 111 with HA[74]
3.1 齐墩果烷型三萜抗冠状病毒的构效关系与作用机制
Wu 等[88]为了鉴定50 种化合物(包括天然产物和合成化合物)是否具有有效的抗SARS 活性,检测了它们对SARS−3CL蛋白酶和病毒进入阶段的抑制活性,发现其中的化合物140 和141(图43)在小于100 μmol/L 的浓度下对SARS−3CL 蛋白酶表现出抑制作用。
为了搜寻更有效的抗SARS−CoV 的化合物,设计了甘草酸的衍生物142 ~156(图44)[89]。构效关系显示,引入2−乙酰胺基−β−D−吡喃葡萄糖胺(化合物142)能大幅度增加其抗病毒活性,部分原因在于引入糖胺可增强化合物的水溶性,另一部分是由于冠状病毒表面蛋白具有高度糖基化的特点,引入糖胺可增强化合物与病毒糖蛋白之间的相互作用。化合物150和152的活性分别较母体提高了45倍和70 倍,具有开发潜力,但细胞毒性较高,大大降低了治疗的安全性;155 和156 的共同特征在于甘草酸C−3 位的糖基被官能团取代,同时C−30 位的羧基被甲酯化,两者均未显示抗SARS−CoV 的活性。说明合理修饰甘草酸可得到更具潜力的抗SARS 冠状病毒衍生物,但是细胞毒性方面也会受到影响,所以要在综合考虑结构修饰和药效的前提下,对甘草酸进行合理改造。
3.2 其他三萜类化合物抗冠状病毒的构效关系与作用机制
化合物157 ~178(图45)是提取自桐叶的天然产物,其中化合物159, 160, 163, 165 ~167, 170 ~172,175 ~178 均对人类冠状病毒HCoV−229E 具有显著的抑制活性,这些结构为开发有效的抗HCoV药物提供了参考[90]。
提取自雷公藤的化合物179 ~182,并利用钯碳将179 还原得到183(图46),5 个化合物均对SARS−CoV 3CL 蛋白酶具有显著的抑制活性,另外,分子对接结果显示,182 C−3 位的羟基可与SARS−CoV 3CL 蛋白酶Cys 44、Cys 44 的羰基的氧原子和Thr 25 的羟基形成氢键(图47),而183 除了疏水相互作用外,没有与酶发生任何键间的相互作用,揭示了C−20 位的甲基对整体构型的影响以及在抑制SARS−CoV 3CL 蛋白酶活性方面的重要作用[91]。这为更深入地研究三萜对于病毒的作用机制提供了参考。
4 三萜抗HBV/HCV 的构效关系与作用机制
乙型肝炎病毒(hepatitis B virus,HBV)和丙型肝炎病毒(hepatitis C virus,HCV)两种病毒都是血源性的,可以通过性接触、从母亲到胎儿的垂直传播以及通过共用针头和输血传播。
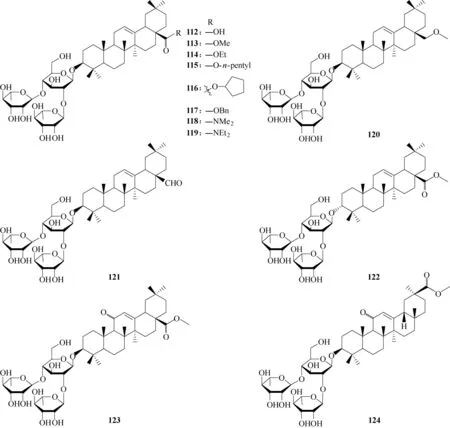
图37 化合物112 ~124的化学结构Fig.37 Chemical structures of compounds 112—124
4.1 三萜抗HBV的构效关系与作用机制
HBV 是一种小型的、具有包膜的双链DNA 病毒,可引起急性和慢性肝炎以及肝硬化。HBV 基因组具有四个开放阅读框(ORF),分别编码x 蛋白、DNA 聚合酶、核心蛋白(包含核心抗原HBcAg 和e抗原HBeAg,HBeAg 埋藏于HBcAg 内部)和表面蛋白(HBsAg),其中HBsAg 由小(SHBs)、中(MHBs)和大(LHBs)三个蛋白组成[92]。HBV 感染的唯一靶标是肝脏细胞,其受体为胆汁酸转运蛋白,即钠牛磺胆酸共转运多肽(sodium taurocholate cotransporting polypeptide,NTCP)[93]。尽管乙型肝炎疫苗的引入已在全球范围内降低了乙型肝炎病毒(HBV)携带者的患病率,但是治疗被慢性HBV 感染的儿童仍然是一个挑战。FDA 批准了几种HBV 抑制剂,如拉米夫定(lamivudine)和阿德福韦(adefovir),然而,严重的副作用和耐药性导致该药物的临床应用受到限制[94],因此,亟需寻找具有新型抗病毒靶标和机制的抗HBV药物。
4.1.1 齐墩果烷型三萜抗HBV 的构效关系与作用机制 甘草酸已用于治疗慢性HBV 感染多年,通过抑制HBV的HBsAg的分泌而发挥抑制作用[95−96]。此外,甘草次酸(glycyrrhetic acid,GA)(图48)已被用作肝靶向药物的配体,并且甘草次酸修饰的载体被证明对肝或肝细胞的靶向递送更为有效[97−98]。基于上述结论,Wang 等[99]研究了一系列甘草次酸衍生物对HBV 的抑制活性,其中化合物184 ~187(图49)对HBV 的DNA 复制阶段表现出明显的抑制作用。特别值得指出的是,化合物187 对于HBV 表面蛋白的抑制能力远强于药物替诺福韦。其构效关系如图50所示。
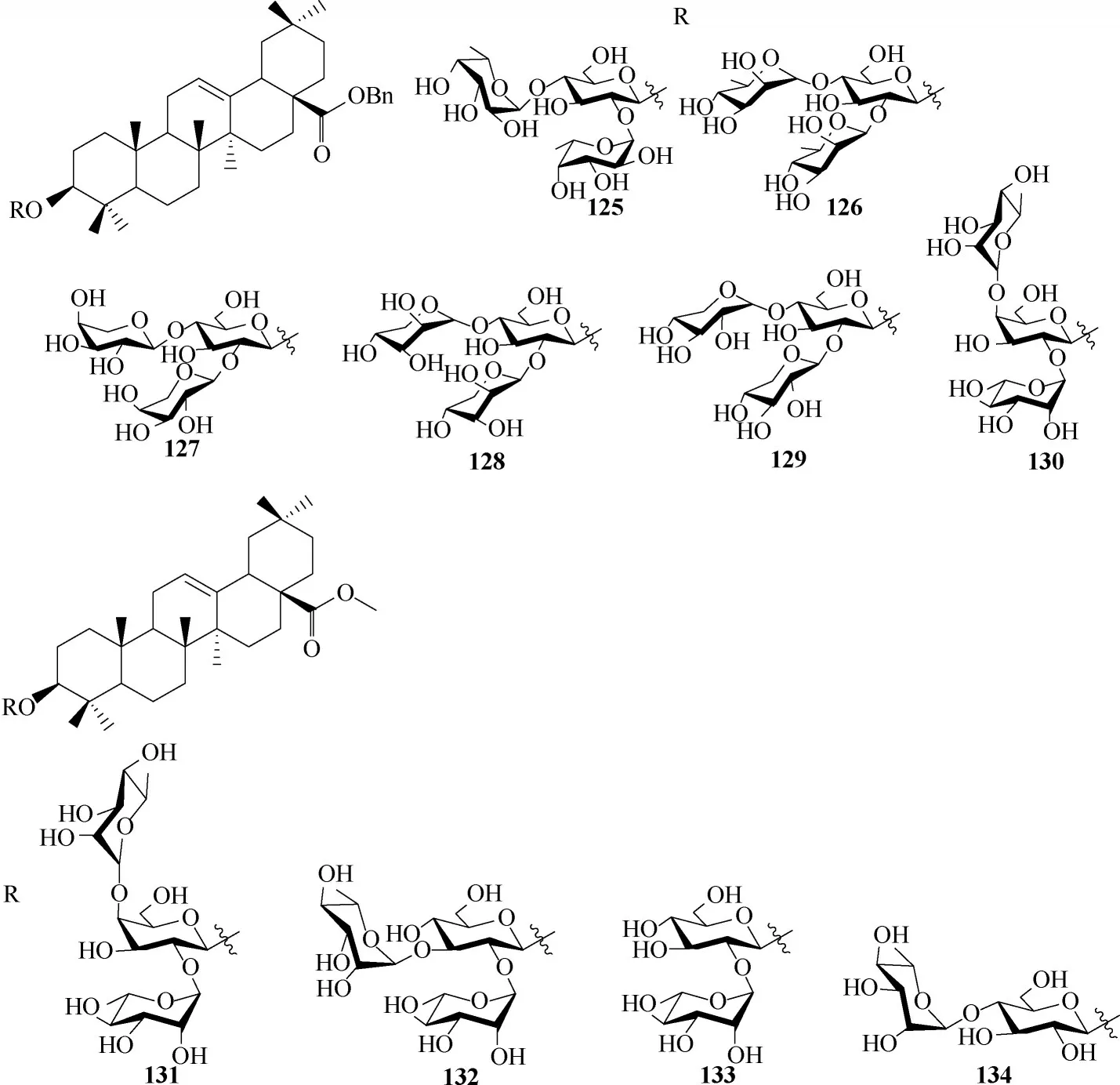
图38 化合物125 ~134的化学结构Fig.38 Chemical structures of compounds 125—134
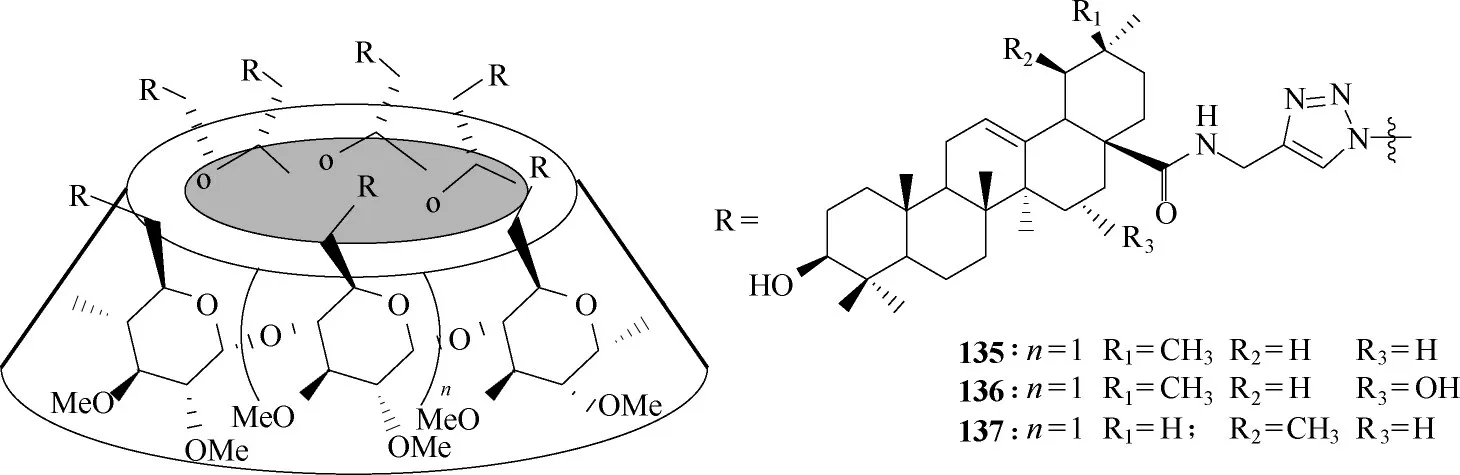
图39 三萜与环糊精缀合物135 ~137的化学结构Fig.39 Chemical structures of conjugates 135—137 of triterpene and cyclodextrin
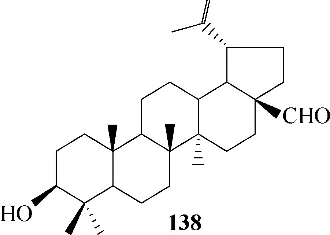
图40 天然产物138的化学结构Fig.40 Chemical structure of natural product 138
4.1.2 羽扇豆烷型三萜抗HBV 的构效关系与作用机制 天然白桦酸及其衍生物已被证明具有良好的抗病毒活性[18]。Yao 等[100]通过转基因小鼠实验证明,在感染HBV 的转基因小鼠的肝细胞中,白桦酸可以抑制含锰超氧化物歧化酶(superoxide dismutase,SOD2)的表达来显著抑制HBV 的复制过程,并且SOD2 的敲低可模仿该抑制作用,而SOD2的过表达则可消除该抑制作用。表明白桦酸介导的HBV 清除是由于线粒体氧化还原平衡的调节所致。SOD2 是位于线粒体中的一种抗氧化酶,可清除超氧阴离子(O2−),避免形成过氧化氢,抑制SOD2表达则导致线粒体中活性氧(ROS)的过度生成,继而可能触发线粒体功能障碍和细胞凋亡。
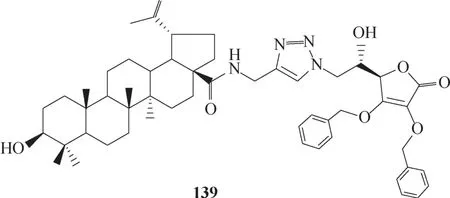
图41 化合物139的化学结构Fig.41 Chemical structure of compound 139
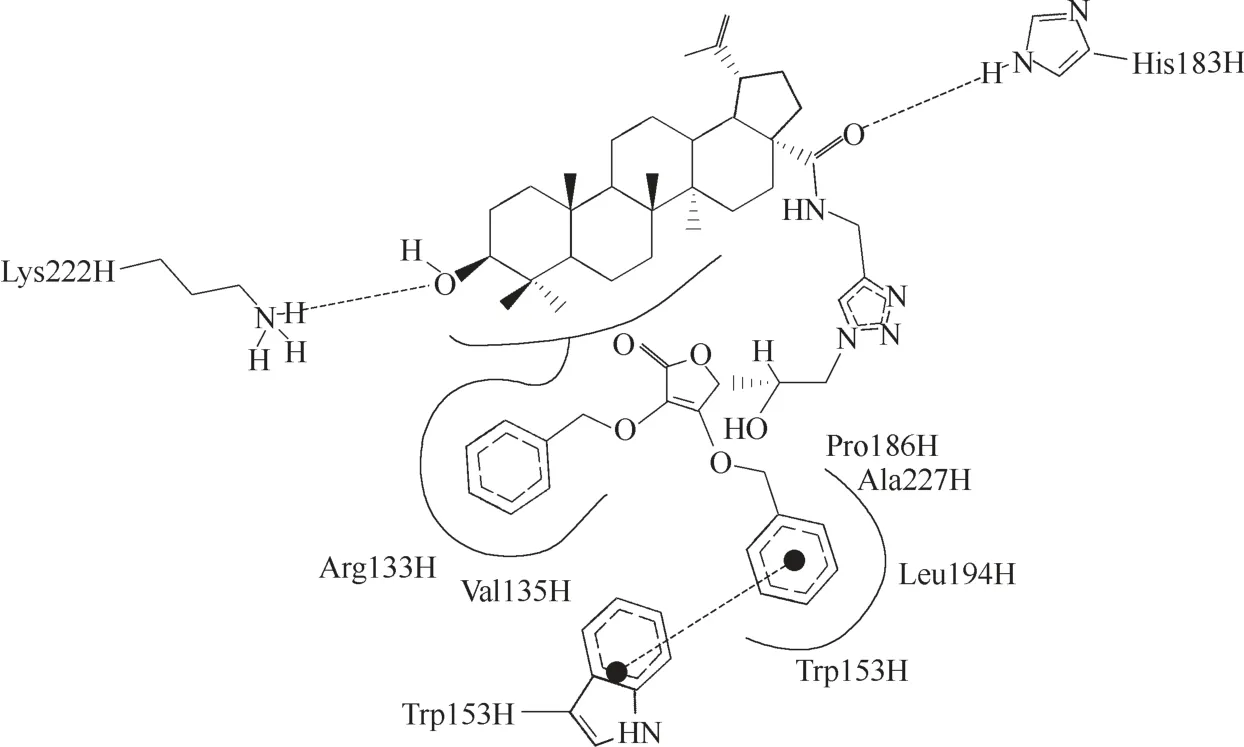
图42 化合物139与HA的对接结果[84]Fig.42 Docking results of compound 139 with HA[84]
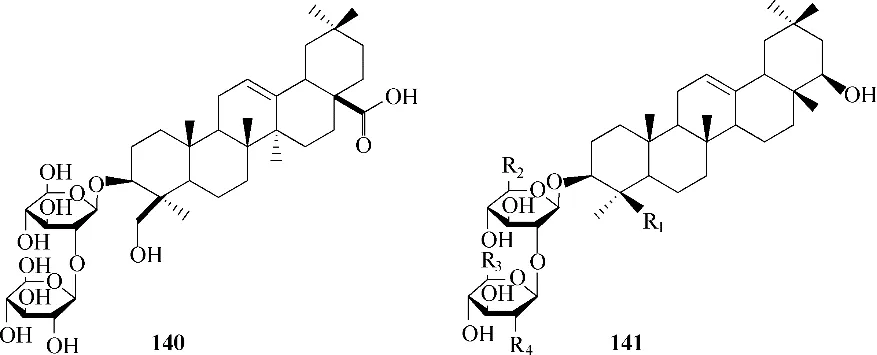
图43 化合物140和141的化学结构Fig.43 Chemical structures of compounds 139 and 140
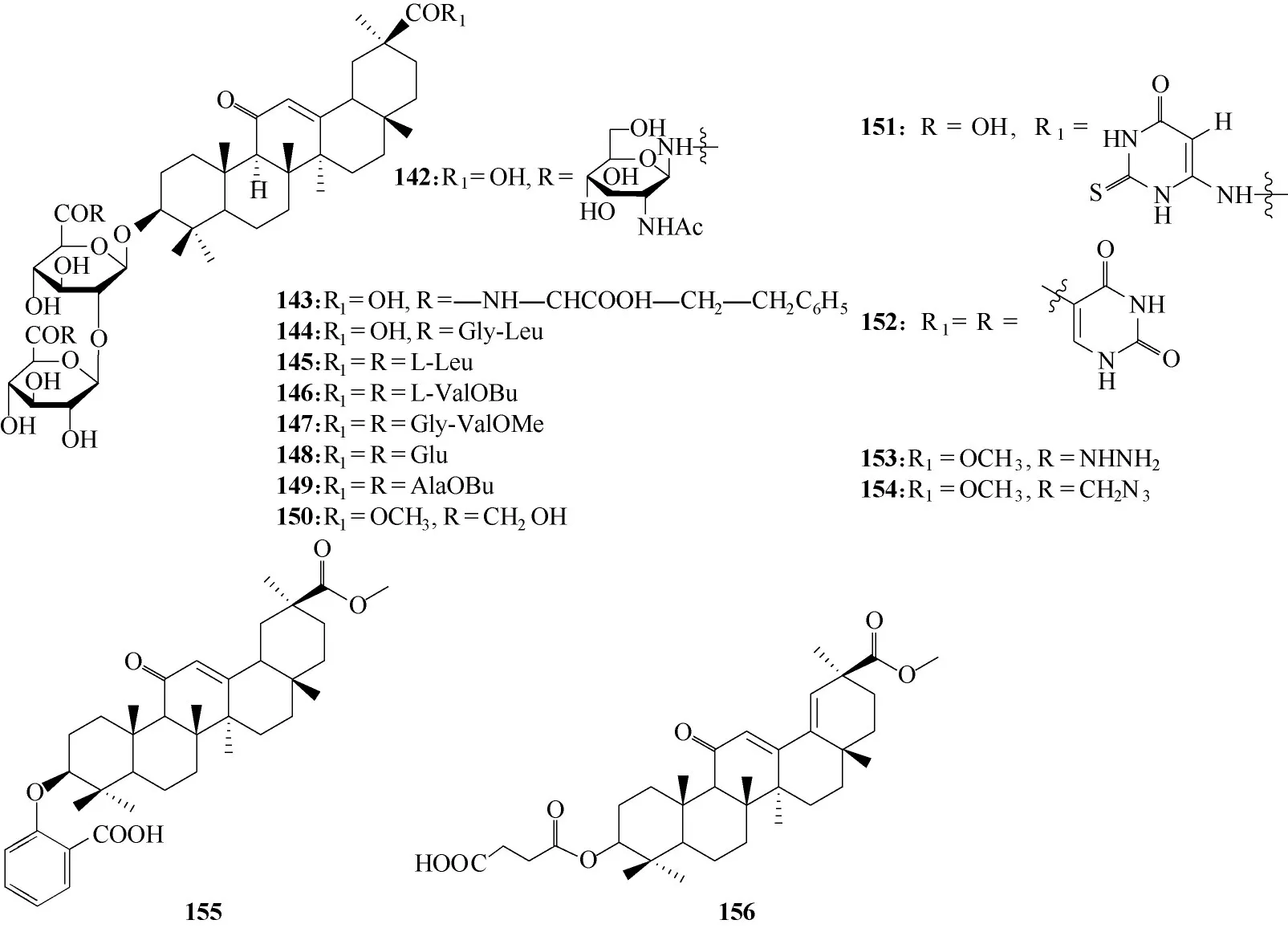
图44 甘草酸衍生物142 ~156的化学结构Fig.44 Chemical structures of glycyrrhizic acid derivatives 142—156
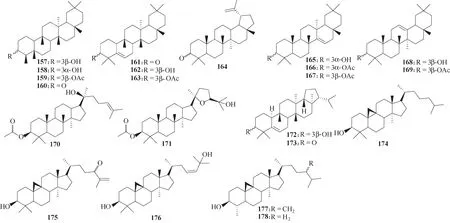
图45 提取自桐叶的天然产物157 ~178的化学结构Fig.45 Chemical structures of natural products 157—178 extracted from the leaves of Euphorbia neriifolia
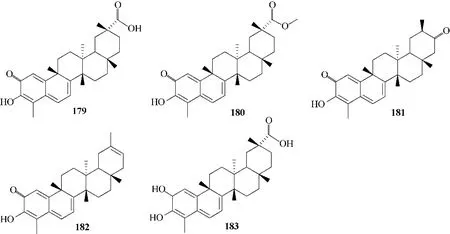
图46 化合物179 ~183的化学结构Fig.46 Chemical structures of compounds 179—183
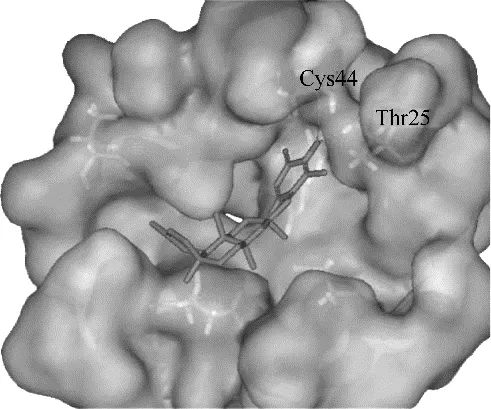
图47 化合物182与SARS−CoV 3CL蛋白酶的对接结果[91]Fig.47 Docking results of compound 182 with SARS−CoV 3CL protease[91]

图48 甘草次酸的化学结构Fig.48 Chemical structure of glycyrrhetinic acid

图49 甘草次酸衍生物184 ~187的化学结构及其抗HBV活性Fig.49 Chemical structures and anti−HBV activity of glycyrrhetinic acid derivatives 184—187
4.1.3 其他三萜抗HBV的构效关系与作用机制提取自西藏蕨麻的天然产物188(图51),被证明可降低细胞中HBV 蛋白HBsAg 和HBeAg 的表达水平,以 及 抑 制HBV 的DNA 复 制[101]。Li 等[102]合 成 了一系列A 环2,3−开环或3,4−内酯的三萜类衍生物(图52,化合物189 ~204),其中190、193、195、198、201、203和204对HBV 蛋白HBsAg和HBeAg的分泌显示出较好的抑制作用;具有C12/C13 双键的193、194、201 和204 的活性比无双键的198、199、202 和203要高,说明C12/C13双键对于维持活性起重要作用;此外,3,4−内酯的水解产物191、194、199 对HBV的DNA 复制的抑制作用略高于190、193、198,表明A 环裂解成游离酸可适当提升化合物对HBV 的DNA复制的抑制活性。
4.2 三萜抗HCV的构效关系与作用机制
HCV 是黄病毒科中的一种具有包膜的正链RNA 病毒,其基因组由5个非翻译区(UTR)、一个开放阅读框和一个短链3′UTR 组成,其中5 个UTR 序列对于病毒的复制必不可少[103]。开放阅读框编码一种多聚蛋白,该蛋白可被宿主和病毒蛋白酶加工成至少10 种成熟病毒蛋白,其顺序依次为NH2-CE1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B-COOH,其中E1和E2是核心结构蛋白,NS5B是一种RNA依赖性RNA 聚合酶(RdRp),E1、E2 和RdRp 是新型抗病毒化合物的有效分子靶标[104]。HCV 是一种血液病毒,随着血液循环流经肝脏时,能特异性地识别肝细胞表面的CD81 受体和一些待确定的辅助受体,从而选择性感染肝细胞,它是导致肝纤维化和肝硬化的主要原因,并最终可导致肝癌[105]。利巴韦林(ribavirin)和聚乙二醇干扰素(pegylated interferon alpha,PEG−IFN−α)联合使用是一种治疗HCV 感染的标准方法,但是由于其带来的副作用使得75%的患者无明显疗效[106]。最近批准的靶向HCV 复制阶段的特拉匹韦(telaprevir)和波普瑞韦(boceprevir)代表了控制HCV 感染的新时代的开始。但是,由于病毒对药物的耐药性,因此仍然亟需找到新型的抗HCV候选药物。
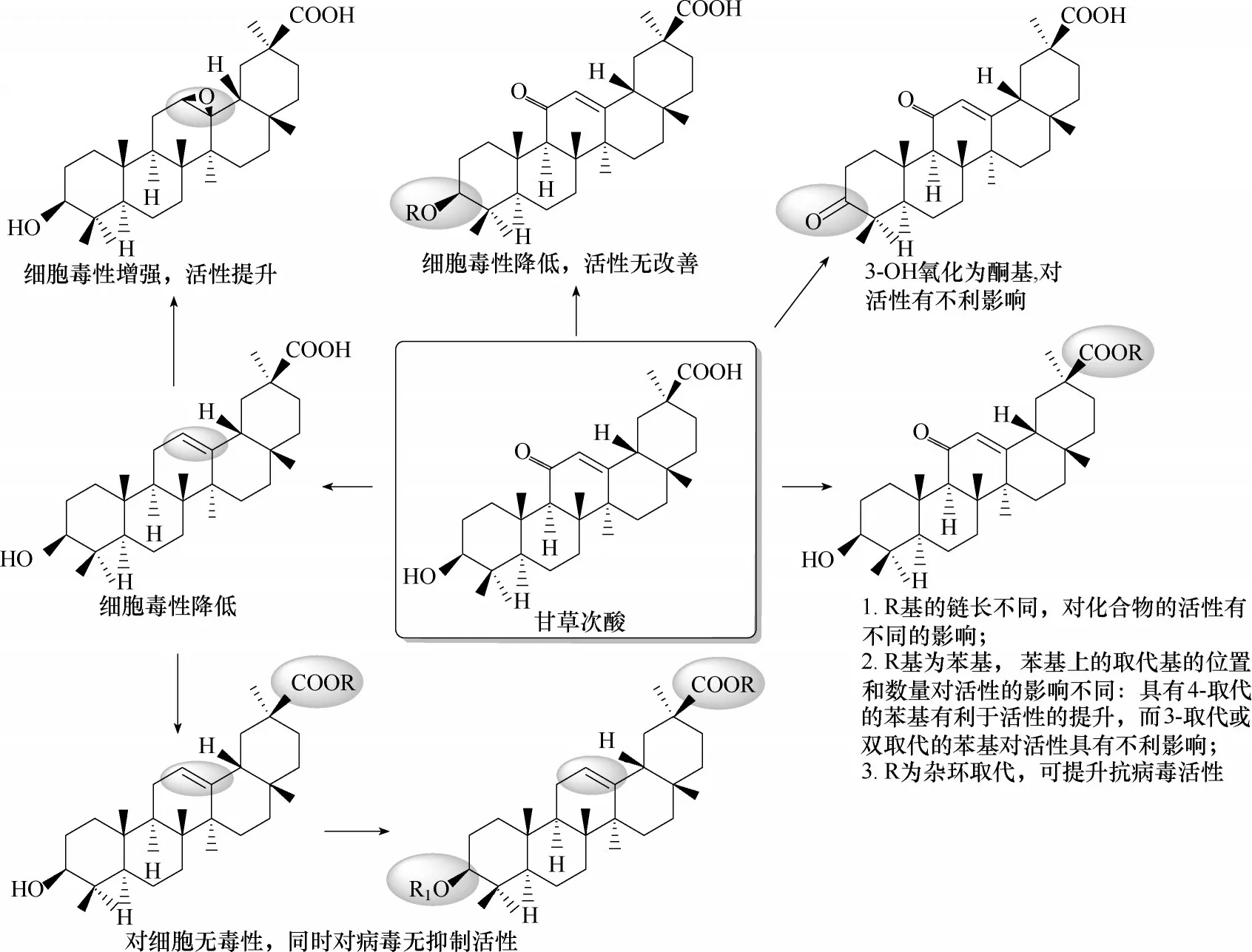
图50 甘草次酸衍生物抗HBV的构效关系Fig.50 Structure−activity relationships of glycyrrhetinic acid derivatives against HBV
图51 天然产物188的化学结构Fig.51 Chemical structure of natural product 188
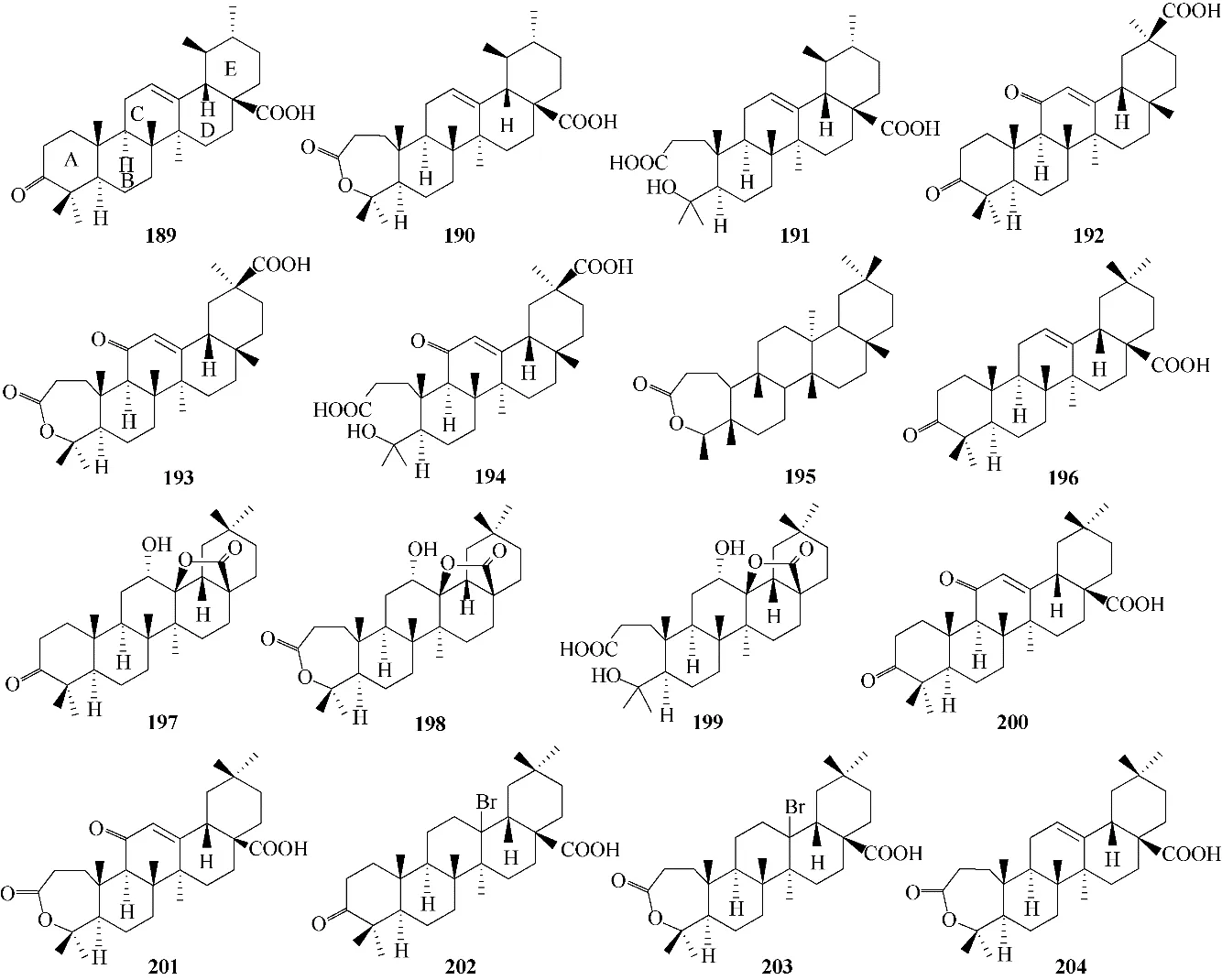
图52 A环为2,3−开环或3,4−内酯的五环三萜类衍生物189 ~204的化学结构Fig.52 Chemical structures of triterpene derivatives 189—204 with seco−A−or 3,4−lactone pentacyclic triterpenoids
4.2.1 甘草酸及其衍生物 提取自甘草干根的甘草酸已被证明能够有效抑制HCV 核心蛋白的表达,且与干扰素具有协同作用,另外,还可减轻由于HCV 引起的肝脂肪变性[107−108]。Matsumoto等[109]已明确甘草酸抑制HCV 的作用机制,即通过抑制具有感染性HCV 颗粒的释放而达到抗病毒效果。大豆皂醇A(soyasapogenol A)(图53)的衍生物ME3738(图2)可抑制HCV 的复制,并与干扰素具有协同作用,表明ME3738 和干扰素的组合可能对慢性丙型肝炎患者有治疗作用[110−111]。
图53 大豆皂醇A的化学结构Fig.53 Chemical structure of soyasapogenol A

图54 化合物205 ~211的化学结构Fig.54 Chemical structures of compounds 205—211
4.2.2 齐墩果酸及其衍生物 据报道,中草药女贞子的水提取物具有直接抑制HCV RdRp 的活性[112]。进一步的分析研究表明,该提取物的两种抗病毒成分为齐墩果酸和熊果酸,两者均可部分地抑制HCV RdRp的活性而达到抗HCV效果[24]。Ma等[113]制备了一系列齐墩果酸衍生物,其中205 ~211(图54)对HCV 蛋白酶具有显著的抑制作用,其构效关系如图55所示,该构效关系对进一步设计和合成作为HCV候选药物的三萜衍生物具有参考意义。
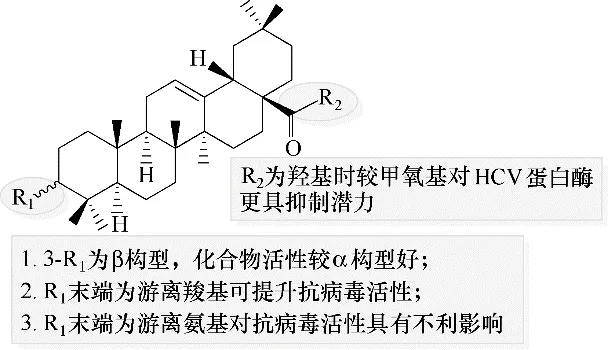
图55 齐墩果酸衍生物抗HCV的构效关系Fig.55 Structure−activity relationships of oleanolic acid derivatives against HCV
图56 刺囊酸的化学结构Fig.56 Chemical structure of echinocystic acid
Yu 等[114−115]的 研 究 显 示,齐 墩 果 酸 和 刺 囊 酸(echinocystic acid,EA)(图56)的衍生物(图57,化合物212 ~227)均表现出良好的抑制HCV进入宿主细胞的活性,它们主要靶点为HCV结构蛋白E2,通过与E2结合而中断E2与其受体CD81之间的相互作用,进而阻断了病毒和宿主细胞的识别过程;此外,此研究还表明刺囊酸的A和B环,以及C、E环的左侧结构是高度保守的,即对其进行修饰或改造,会对抗病毒活性有不利影响,而对D环进行合理修饰可提高抗病毒活性,并由此推定了刺囊酸及其衍生物抑制HCV进入宿主细胞的作用机制(图58)。Wang等[116]通过开环或扩环实验进一步证明了以上结论。
然而,HCV 结构蛋白与其受体CD81 之间存在至少两个口袋,对于刺囊酸这样的小分子很难通过竞争作用来占据口袋,因此在进一步研究中,Yu等[114,117]试图将多个刺囊酸分子偶联在一起,以增强药效。通过对连接子进行一系列改进和优化,得到的二聚物228 ~232(图59)具有较好的抗HCV 活性,其中化合物228 和231 最具潜力。另外,刺囊酸二聚体的刺囊酸部分高度保守,任何修饰或取代都可能显著降低甚至消除其抗HCV活性[118]。

图57 齐墩果酸与刺囊酸衍生物的化学结构(212 ~224为齐墩果酸的衍生物,225 ~227为刺囊酸的衍生物)Fig.57 Chemical structures of oleanolic acid derivatives and echinocystic acid derivatives(212—224 are oleanolic acid derivatives,225—227 are echinocystic acid derivatives)
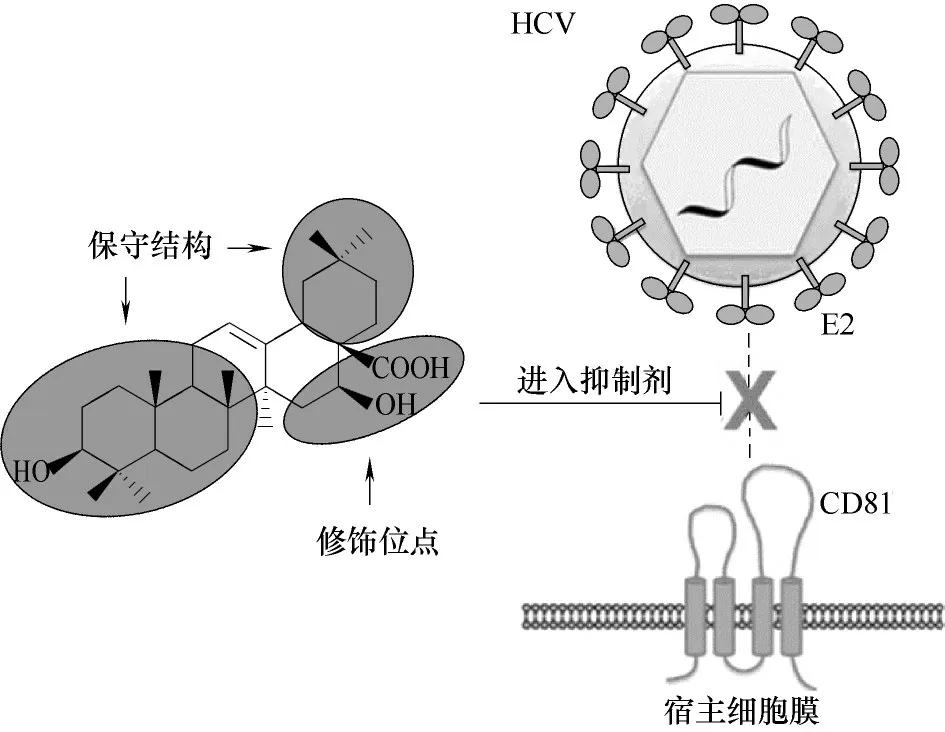
图58 推定的刺囊酸及其衍生物抗HCV的作用机制[114](经ACS出版社授权)Fig.58 A proposed mechanism for echinocystic acid and its derivatives against HCV[114](With the permission of ACS Press)
即便三萜类化合物可有效抑制HCV 进入宿主细胞,但还是存在水溶性较低的问题,这将导致该类药物的生物利用度低。而将三萜与环糊精偶联所得的缀合物233 ~252(图60),其水溶性得到改善,并且大部分可维持或提高其抗HCV 活性。最具潜力的化合物248 和251,平均IC50值(抑制50%HCV 复制所需的浓度)分别为1.18 μmol/L 和0.25 μmol/L,并且在浓度为100 μmol/L时,未观察到其对细胞产生的毒性作用[119−120]。此研究为开发三萜类缀合物作为HCV 进入阶段的抑制剂提供了理论依据。
5 结论与展望

图59 化合物228 ~232的化学结构Fig.59 Chemical structures of compounds 228—232
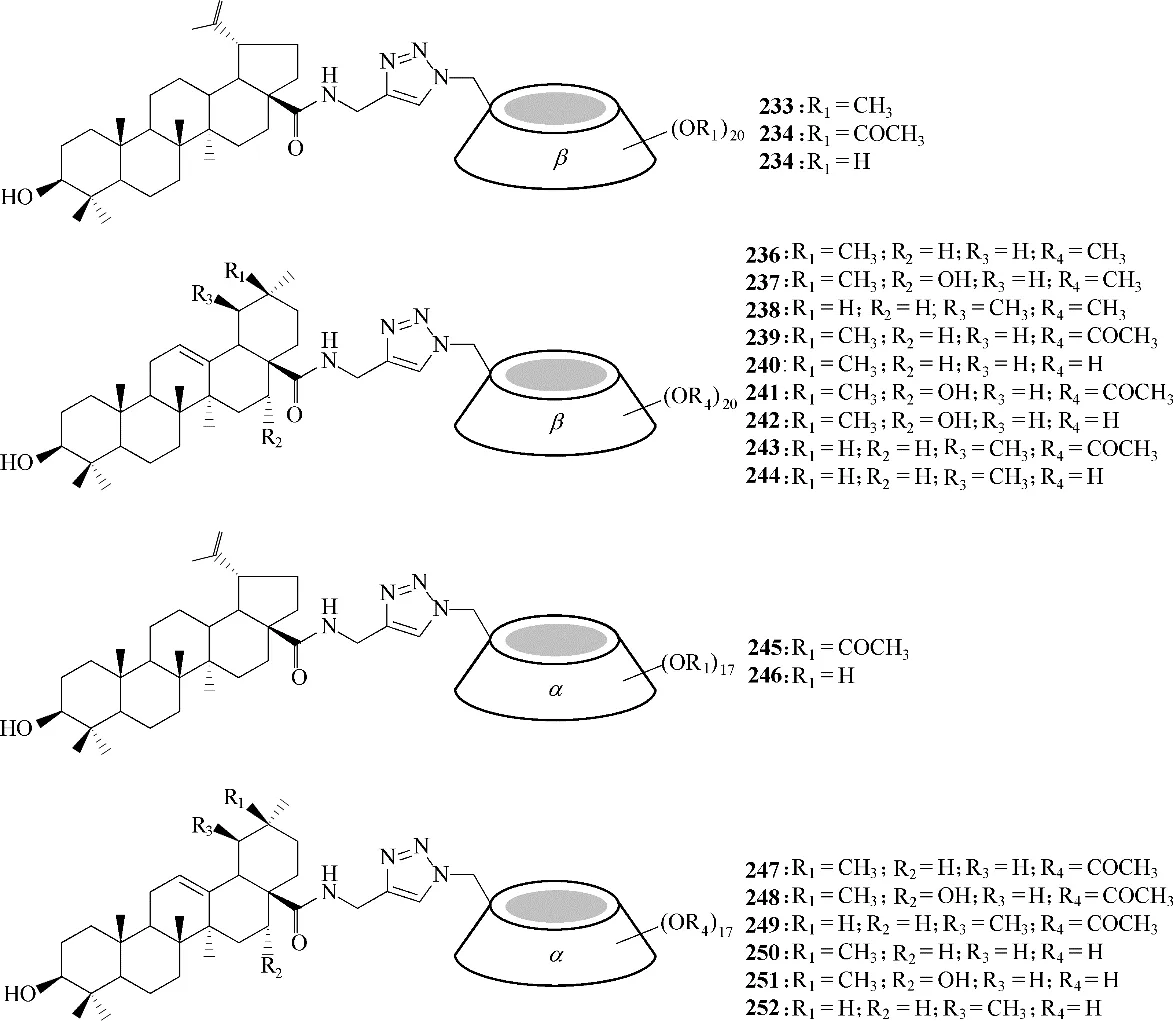
图60 三萜与环糊精的缀合物233 ~252的化学结构Fig.60 Chemical structures of conjugates 233—252 of triterpene and cyclodextrin
研究表明许多天然三萜及其衍生物除具有抗HIV、流感病毒、冠状病毒、HBV/HCV 活性外,对抗单纯疱疹病毒[121−123]、人呼吸道合胞病毒[124−125]、寨卡病毒[126]、乳头状瘤病毒[127−128]等病毒也具有抑制活性。三萜骨架目前主要靠植物萃取分离来获取,来源受到限制,三萜在预防和治疗病毒性疾病中的价值尚未被完全开发,只有极少数三萜化合物进入到IIb 期临床试验。由于对抑制剂的确切靶标和作用机制尚不明确,同时很多化合物水溶性较差,所以限制了它们的成药性。因此仍然需要投入大量的时间和精力对其进行更加深入的探索,包括:(1)借助合成生物学和计算机虚拟筛选技术发掘更多的三萜骨架结构,并解决科学研究的先导化合物和工业生产的原料药的来源问题;(2)借助有机化学和药学阐明构效关系和作用靶标,以期基于生物大分子结构或者药效团模型合理设计出具有高效抗病毒活性制剂,同时通过结构修饰增强此类化合物与靶标的结合力,以及改善其成药性,如引入合适的非天然氨基酸或含有酰胺基团的小分子。
