二维金属有机框架及其衍生物用于电催化分解水的研究进展
2020-09-29马佳欢杨微微白羽孙克宁
马佳欢,杨微微,白羽,孙克宁
(北京理工大学北京市化学电源与绿色催化重点实验室,北京100081)
引 言
随着世界人口的不断增长和经济的快速发展,全球对能源的需求和对环境污染的治理压力日益增大,因此世界各国科学家开始关注绿色的、可再生的新型能源。氢气是一种能量密度较高(140.4 MJ·kg−1)的清洁能源,被认为是化石能源的理想替代品[1]。电催化水分解是目前最有效的制氢方法之一,具有成本低、效率高、产物无污染、来源丰富等优点,且其需要的电能可由太阳能、风能等清洁的可再生能源转化而来。电催化水分解包括两个半反应,即阴极析氢反应(HER)和阳极析氧反应(OER),如图1(a)所示[2]。考虑到发生水分解的电解质不同,水分解反应可表示如下:
总反应:
在酸性电解质中:
阴极:
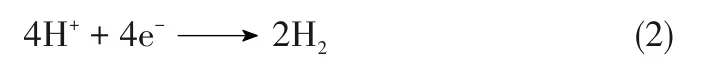
阳极:
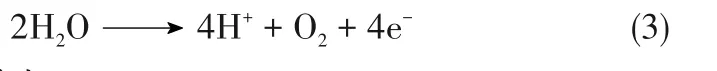
在碱性电解质中:
阴极:
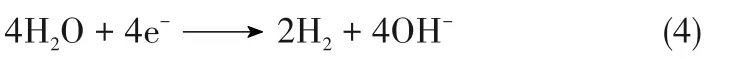
阳极:
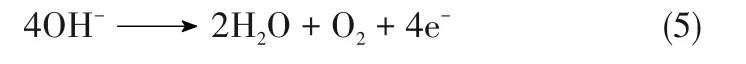
HER 反应是二电子转移过程,通常在酸性介质中的反应机制包括三个步骤[3−4]:首先是Volmer 步骤(H++ e−→H*),电子与质子在电极表面反应产生一个被吸附的氢原子(H*),其中*代表催化剂活性位点。产生H*后,通过Heyrovsky(H* + H++ e−→H2)和Tafel(H*+H*→H2)两个步骤竞争或同时进行H2析出反应。当催化剂表面形成的H*相对较少时,依次发生Volmer−Heyrovsky 步骤[图1(b)中的右侧路线]。但是,当催化剂上生成的H*足够多时,将进行更快的Volmer−Tafel 步骤[图1(b)中的左侧路线]。研究表明H*在催化剂表面的吸附Gibbs 自由能(ΔGH*)影响HER 反应效率,通常将ΔGH*值接近零的材料视为良好的HER 催化剂。在碱性条件下需要经过水解离步骤形成H*(H2O+e−→H*+OH−),因此催化剂的HER 活性同时受到水解离步骤和ΔGH*的影响[5−8]。Chen 等[8]研究发现在NiFe−LDH 中掺杂Ru 原子后,其ΔGH*几乎没有发生变化,但是水解离能从1.02 eV 降至0.5 eV。此时材料催化HER的活性显著提升,在1 mol·L−1KOH 水溶液中,电流密度10 mA·cm−2下所需过电势从269 mV(NiFe−LDH)降低至29 mV(NiFeRu−LDH)。这说明碱性条件下的水解离步骤对于HER 反应动力学具有显著的影响。
OER 是四电子转移过程,所需要的过电势较大,能量转化效率较低,是电催化水分解的关键步骤。在碱性条件下,首先OH−→OH* + e−,OH* +OH−→O* + H2O + e−,随后氧气析出步骤可分为两类:如图1(c)所示,绿色箭头表示从O*中间体直接析出氧气的路径(O* + O* →O2),黑色圆圈表示随后产生OOH*和O2的路线(O* + OH−→OOH* + e−,OOH*+ OH−→O2+H2O + e−)[9]。在酸性介质中OER反应过程与碱性条件类似,都涉及反应中间体OH*、O*和OOH*,所以人们普遍认为OER活性很大程度上取决于中间体的表面结合能[10−11]。目前商业化的贵金属催化剂主要是IrO2和RuO2,其中,IrO2与氧中间体的结合力太强,而RuO2的结合力较弱,因此这两种催化剂都不满足最佳的OER 条件[12]。为了实现大规模的应用,电催化剂应具有催化活性高、循环稳定性好、成本低、储量丰富、易合成等特点。近年来,研究人员致力于开发能有效促进制氢技术发展的非贵金属电催化剂。
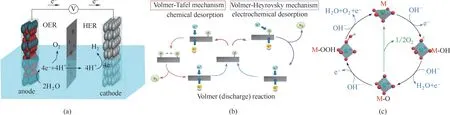
图1 (a)电化学水分解系统的示意图[2];(b)酸性溶液中电极表面析氢的两种机理[4];(c)碱性溶液中电极表面的析氧机理[11]Fig.1 (a)Schematic diagram of an electrochemical water splitting system[2];(b)The two mechanism of hydrogen evolution on the surface of an electrode in acidic solutions[4];(c)The mechanism of oxygen evolution on the surface of an electrode in alkaline solutions[11]
金属有机框架(metal−organic framework,MOFs)材料是由金属离子节点和有机配体通过配位作用自组装形成的一类多孔晶体材料[13−14],兼具无机材料的高结晶度和有机材料的高比表面积、高孔隙率等优点,被广泛应用于电催化领域[15−18]。然而,MOFs通常以三维网络结构的形式存在,金属离子中心具有较大的配位数,所以导电性较差的有机配体致使大多数MOFs 的电导率较低(约10−10S·m−1)[19]。同时,三维结构MOFs 内部的曲折通道阻碍了活性位点的暴露和物质的传输,降低了活性中心与反应物的接触概率,导致催化活性的降低。通过调控尺寸和形貌获得低维度的MOFs 晶体可以有效增大MOFs 材料的比表面积,暴露出更多的活性位点,从而有效提高材料的电催化性能[20−22]。
近年来二维MOFs 由于其独特的结构优势被广泛应用于电催化分解水中[23−25]。与三维MOFs 相比,二维MOFs 纳米片具有较大的横向尺寸、大的比表面积和丰富的表面不饱和金属活性位点,有利于反应物的吸附和活性位点的暴露[26]。并且当二维MOFs 纳米片的厚度降至纳米级别时,电导率会大幅增加,从而有利于其电催化性能的提升[27]。此外,以二维MOFs 为前体/模板还可以获得金属化合物−碳基材料,碳层的形成不仅提高了材料的导电性,而且有效减缓了金属化合物纳米粒子在电解液中的腐蚀,提高了催化剂的稳定性[28]。
本文从组分调节和结构调控两方面介绍了二维MOFs 材料及其衍生物在电催化水分解领域的最新研究进展,最后提出二维MOFs 材料及其衍生物作为电催化剂存在的挑战及未来的发展前景。
1 二维MOFs用于电催化水分解
二维MOFs 纳米片的合成方法主要有自上向下和自下向上两种策略。自上向下方法是将三维层状结构的MOFs 剥离,而自下向上法是指由金属中心和有机配体直接合成二维MOFs 纳米片。层状结构的MOFs 在层内具有较强的配位键,而层间的范德华力或氢键较弱,自上向下的方法可以克服层间弱相互作用,使层状结构的MOFs 发生剥离,形成二维MOFs 纳米片[29]。自下向上的方法则是通过在一个方向上限制配位聚合物的生长,或者在合成过程中限制/抑制晶体层间的相互作用来制备二维MOFs[26,30−31],一些自下向上的方法甚至可以用来制备二维非层状结构的MOFs 纳米片,如UiO−67 纳米片[32]、OX−1 纳米片[33]、ZIF−8 纳米片[34]和ZIF−67 纳米片[35]。
二维MOFs 的大比表面积、高暴露活性位点等特性使其具有优异的电催化活性[16]。研究表明,调节二维MOFs 的金属中心和配体组分能优化催化剂电子构型,促进限速步骤中间体的吸附,实现更快的电子转移,从而提高材料的本征活性[36]。此外,通过进一步的复合结构调控将二维MOFs 生长在各种基底上或与其他材料复合能有效增强其电催化水分解性能。
1.1 金属中心调控
1.1.1 单金属二维MOFs 过渡金属(Fe、Co、Ni 等)离子和丰富的有机配体通过配位作用可以组装孔径、组成和厚度可调的多种二维MOFs。由于二维MOFs 具有高表面积和大量暴露的金属活性中心,是优异的水分解电催化剂。
Jayaramulu 等[37]通过机械研磨硝酸钴和苯并咪唑合成本体ZIF−9(Ⅲ),随后通过超声辅助液相剥离法将大块MOF 剥离成超薄二维钴基ZIF−9(Ⅲ)纳米片,如图2(a)、(b)所示。该纳米片催化剂在碱性条件下表现出优异的OER 活性,在10 mA·cm−2的电流密度下仅需0.38 V 的过电势,远低于相应的本体ZIF−9(Ⅲ)。二维ZIF−9(Ⅲ)纳米片具有高的OER 活性主要因为以下两方面:(1)与氮配位的高密度羟基氧化钴(N4CoOOH)活性位点具有高度的可接触性,以及与Co 配位的N 原子通过影响Co 位点的电子性能,改变反应中间体的吸附活性;(2)二维MOF 的纳米片形貌使底物分子具有较小的扩散障碍,从而导致剥落的2D ZIF−9(Ⅲ)纳米片具有较强的OER 活性。Huang等[38]利用电化学/化学剥离的方法将三维MOF 原位剥离成超薄二维Co−MOF 纳米片,如图2(c)~(e)所示。当3D 柱状层结构MOF 用作水氧化的电催化剂(pH=13)时,柱状配体被原位氧化并去除,形成了超薄(2 nm)的二维Co−MOF 纳米片。与3D柱状层结构MOF 相比,剥离得到的二维超薄纳米片的表面原子和活性位点能够更加充分暴露,具有低过电势、大电流密度和高TOF 值,展示出高的OER活性。
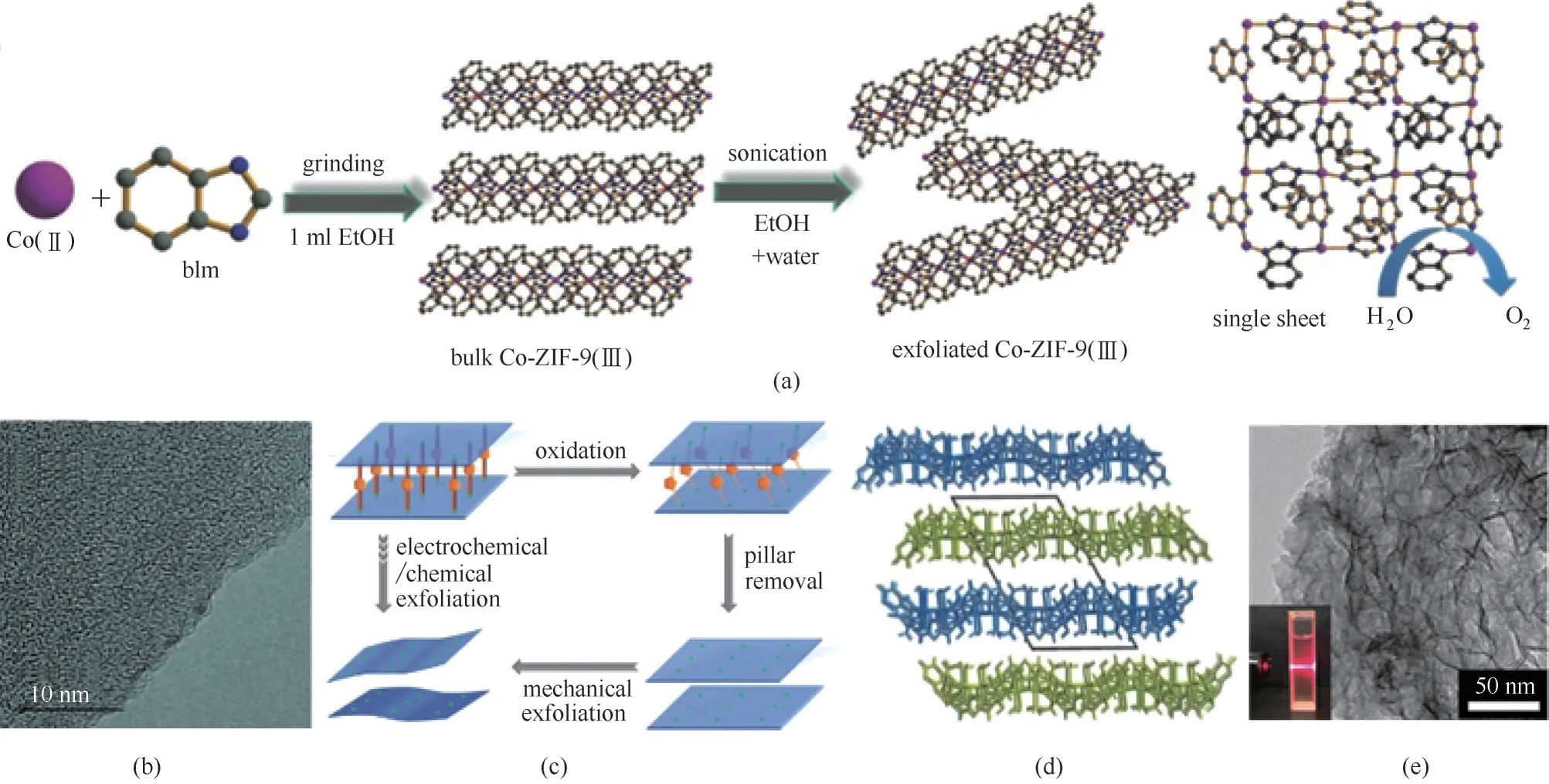
图2 (a)二维Co−ZIF−9(Ⅲ)纳米片在液相中从块状Co−ZIF−9(Ⅲ)剥离的示意图;(b)液相剥离的2D超薄纳米薄片Co−ZIF−9(Ⅲ)的HAADF−TEM 图[37];(c)柱状MOF的选择性剥离示意图;(d)在2D−Co−NS中沿b轴看的2D层结构,交替层用蓝色和绿色区分;(e)2D−Co−NS的TEM图像(插图:胶体溶液的丁达尔效应)[38]Fig.2 (a)Schematic representation of 2D Co−ZIF−9(Ⅲ)nanosheets exfoliated in liquid phase from bulk Co−ZIF−9(Ⅲ);(b)HAADF−TEM image of liquid exfoliated 2D ZIF−9(Ⅲ)showing 2D ultrathin nanosheets interactions[37];(c)Selective pillar removal and exfoliation of a pillared−layer MOF;(d)3D packing structure of the 2D layers viewing along the b−axis in 2D−Co,alternate layers blue and green for clarity;(e)A TEM image of 2D−Co−NS(inset:Tyndall effect of a colloidal solution) [38]
Xu 等[39]通过简单的表面活性剂辅助水热法将Co2+和苯二甲酸合成了超薄二维Co−MOF 纳米片。在微观结构上,Co原子与6个O原子配位,形成的伪八面体在(200)晶体平面上沿[010]/[001]方向进一步相互连接,形成由配体BDC 分子分隔的紧密堆积的金属原子平面(200),即最大的暴露表面。所获得的产物表现出优异的OER 电催化活性,在1 mol·L−1KOH 溶液中,当电流密度为10 mA·cm−2时仅需263 mV 的低过电势,并且显示出74 mV·dec−1的小Tafel斜率。优异的电催化性能主要归因于二维Co−MOF纳米片超薄的厚度和表面暴露的不饱和CoⅡ活性位点:超薄厚度有利于活性位点与底物分子之间的更多相互作用,从而改善电催化性能;OER 催化过程中CoⅡ活性位点被部分氧化为更高价态,提高了材料的OER 催化活性。同时MOF 材料的多孔结构还提供了丰富的扩散通道,有助于快速的物质传输和电子转移,从而提高反应动力学。
Jia 等[40]采用自下向上的方法,首次设计并合成了一种新型的酞菁镍基二维MOF(NiPc−MOF),合成步骤及结构如图3所示。通过多种光谱表征手段验证了NiPc−MOF 具有四重对称性的大π 共轭结构。基于其高比表面积、分级多孔结构和良好的导电性,NiPc−MOF 材料被直接用于电催化水分解。NiPc 基序不仅作为连接单元,而且充当电催化活性位点,一系列电化学测试证明其在碱性条件下具有杰出的OER性能。
Song 等[41]通过自上向下的策略合成了由一维[Co4(OH)2]6+链组成的超薄CoII−有机纳米片,如图4(a)所示。首先通过Co(OH)2和戊二酸的水热反应生成3D TMOF−4,之后在高温下用含有过量十二烷基硫酸钠(SDS)的水溶液将TMOF−4 处理12 h。由于配体交换过程的强烈诱导,过量的SDS 阴离子部分取代了谷氨酸盐,同时,单齿SDS使相邻金属有机层之间减少连通性,从而导致超薄二维纳米片的形成,其晶体结构如图4(b)、(c)所示。与剥落前的块状MOF相比,具有原子厚度的超薄MOF纳米片结构显示出更优异的OER 催化活性,如图4(d)所示,在10 mA·cm−2电流密度下的过电势为318 mV,Tafel 斜率为54 mV·dec−1。电化学研究表明,金属有机纳米片的高OER 活性来自催化活性位点CoII的高度可及性和1D钴酸盐链的高电荷转移能力。
1.1.2 多元金属二维MOFs 双金属二维MOFs 具有优异的电催化性能,在单金属MOFs 中引入第二种金属元素能调节材料原有的电子结构,提高电子转移速率,优化本征催化活性。最近基于双金属二维MOFs工作的电催化性能总结在表1中。
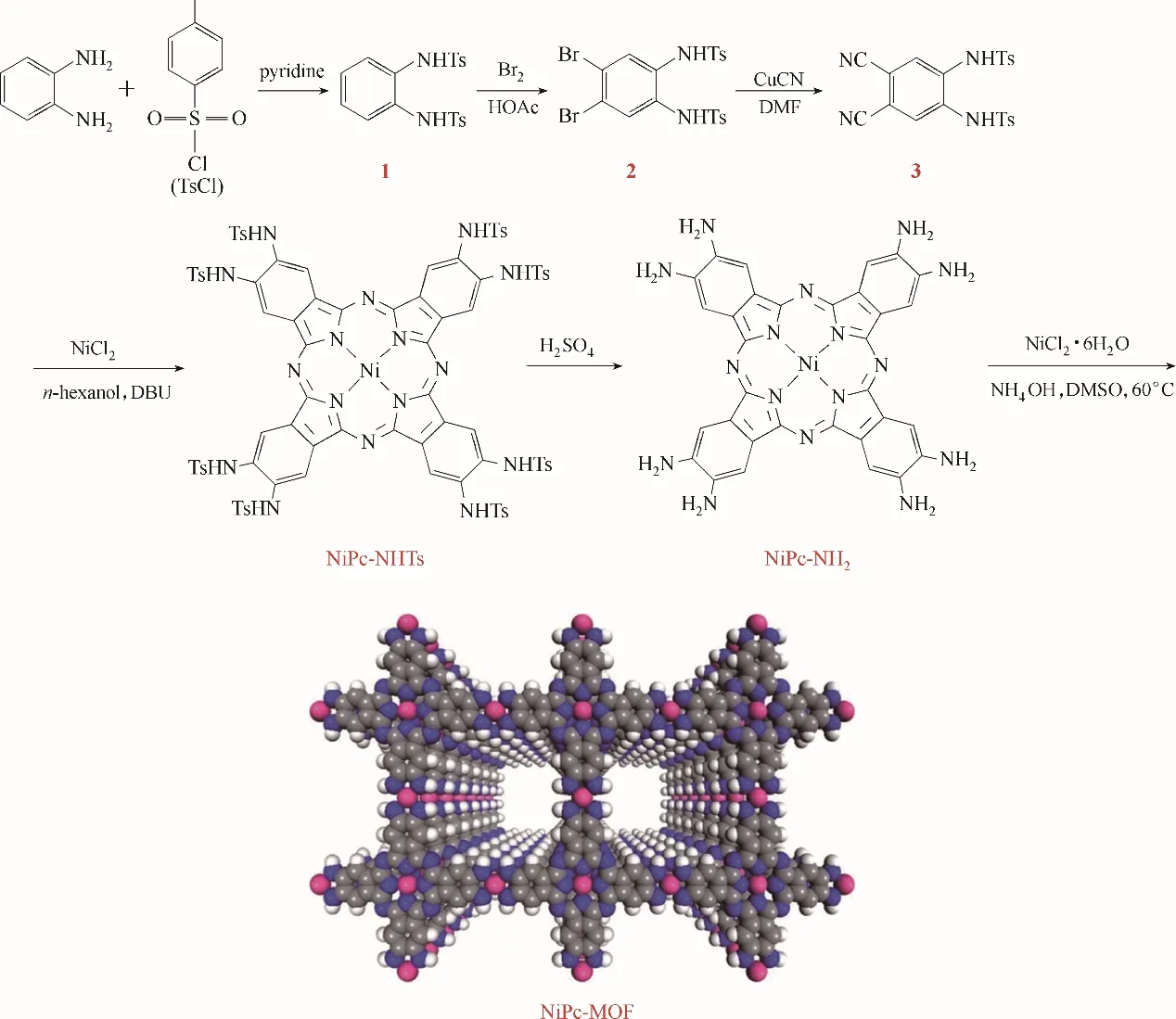
图3 NiPc−MOF的化学合成步骤及其化学结构[40]Fig.3 Chemical synthesis procedure for the NiPc−MOF and its chemical structure[40]
Zhao 等[42]通过超声辅助的水热法,合成了厚度仅 为3.1 nm 的 双 金 属MOFs 纳 米 片(NiCo−UMOFNs),3.1 nm 的均匀厚度对应于4 个金属配位层或3 个配位结构层(2.9 nm),金属原子在(200)平面上呈六角形排列,如图5(a)~(c)所示。NiCo−UMOFNs 厚度决定了暴露的活性表面的百分比,超薄厚度使得表面存在配位不饱和金属活性位点,且Ni2+和Co2+耦合后,O2−与Ni2+之间的电子−电子排斥作用会增强Co−O 的π 电子云密度,促进电子从Ni2+向Co2+转移,使得双金属二维MOFs片层具有优异的OER性能。在1 mol·L−1KOH电解质溶液中,电流密度为10 mA·cm−2时,NiCo−UMOFNs 仅需250 mV 的过 电 势,催 化 活 性 远 高 于Ni−UMOFNs 和Co−UMOFNs 的机械混合物。Pang 等[43]采用物理超声和化学溶剂作用相结合的方法,利用H2O 作为替代分子交换柱状配体,将三维柱状层MOFs 剥离成二维层状MOFs纳米片,如图5(d)所示。将二维CoNi(bdc)用作OER 反应催化剂时,在中性电解质中,1 mA·cm−2时过电势为344 mV,如图5(e)所示,且在1.8 V电压下电解16 h 后,活性几乎没有损失,表明其具有良好的电化学稳定性。
Li 等[45]开发了一种大规模的、自下向上的溶剂热法制备Ni−M−MOF(M = Fe,Al,Co,Mn,Zn 和Cd)超薄纳米片。纳米片边缘略微卷曲,厚度为1.67~2.58 nm,如图6(a)、(b)所示。在1 mol·L−1KOH 电解质溶液中,双金属Ni−Fe−MOF 纳米片表现出优异的OER 性能,电流密度为10 mA·cm−2时,过电位仅为221 mV,测试20 h 电流密度没有显著的损失。通过DFT计算和实验说明镍原子和铁原子之间存在强相互作用,引入铁原子后Ni2+被氧化成Ni3+,增强了电子电导率和本征活性,有效改善双金属Ni−Fe−MOF NSs的OER催化活性。

图4 (a)TMOF−4和相应的纳米片的合成过程示意图;(b)TMOF−4沿a轴的晶体学视图;(c)沿b轴的单个[Co4(OH)2]6+的晶体学视图(Co:橙色,O:红色,C:黑色);(d)TMOF−4纳米片的低放大TEM图像和LSV极化曲线[41]Fig.4 (a)Schematic illustration of the synthesis process for TMOF−4 and the corresponding naonsheets;(b)crystallographic view of TMOF−4 along the a−axis;(c)crystallographic view of a single[Co4(OH)2]6+along the b−axis(Co:orange,O:red,C:black);(d)low−magnification TEM image and LSV polarization curves of TMOF−4 nanosheets[41]
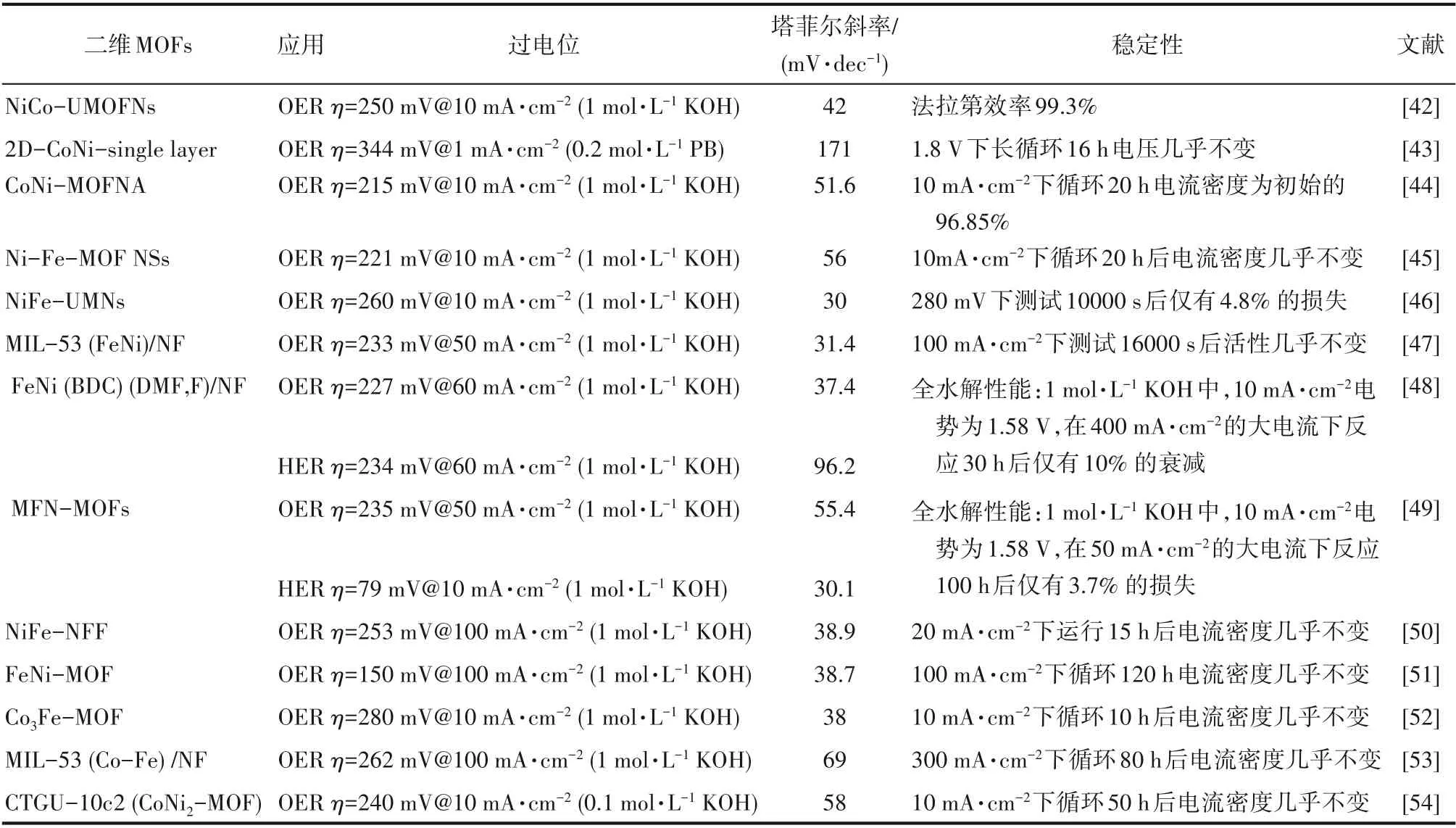
表1 二维双金属MOFs在电解水中的应用Table 1 2D bimetallic MOFs for electrocatalytic water splitting
Hai 等[46]采用自下向上溶剂热及超声辅助的方法制备了厚度为10 nm左右双金属超薄Ni−Fe−MOF纳米片(NiFe−UMNs),如图6(c)所示。在O2饱和的1 mol·L−1KOH电解质溶液中,表现出优异的OER性能,在10 mA·cm−2时过电位为260 mV。研究表明NiFe−UMNs 表面暴露出大量配位不饱和活性金属位点。XPS 与DFT 计算结果表明Ni 和Fe 的结合可以优化eg轨道的填充进而提高OER 活性。Sun 等[47]在泡沫镍表面合成MIL−53(FeNi)/NF。FT−IR 结果显示MIL−53(FeNi)表面有亲水性羧酸基团存在,有利于促进水分子的吸附,同时,在MIL−53(Ni)结构中引入Fe原子能增强反应物的吸附。此外,部分不稳定三维态和增加的三维轨道电子密度有利于中间体的反应,加速催化过程。Lin 等[48]也在泡沫镍基底上制备了FeNi(BDC)(DMF,F)/NF 材料,配位方式如图6(d)、(e)所示,表明DMF 分子与只有Fe 和Fe 丰富的MOFs 的核心金属离子配位,而与只有Ni 和Ni 丰富的MOFs不存在配位。双金属MOFs/NF 电极表现出杰出的OER和HER性能,进一步将其作为双功能电催化剂在1 mol·L−1KOH 的双电极体系中进行整体水分解,仅需1.58 V 即可达到10 mA·cm−2的电流密度,而且在400 mA·cm−2的大电流下反应30 h 后仅有10%的衰减。
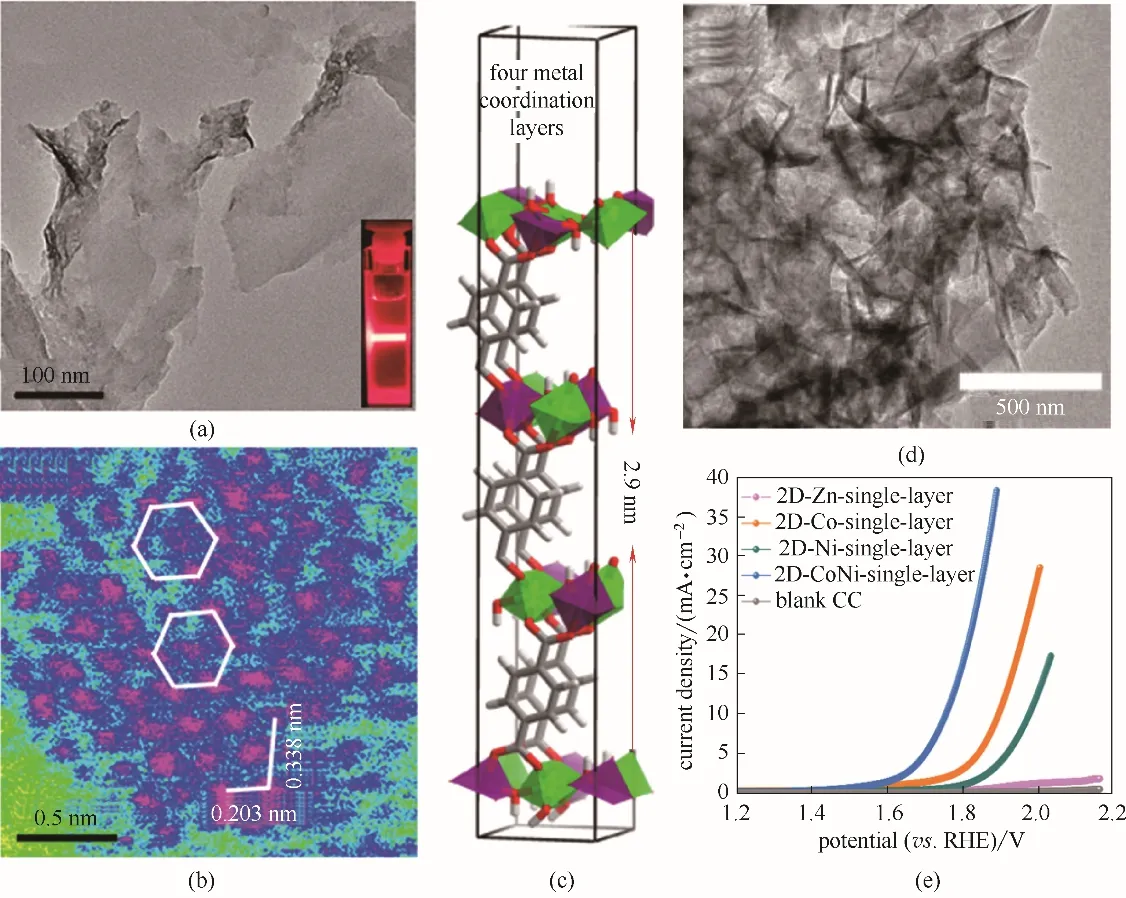
图5 (a)NiCo−UMOFNs的TEM图像,插图显示了NiCo−UMOFNs在水溶液中的丁达尔效应;(b)NiCo−UMOFNs(200)平面的HAADF−STEM 图像,粉红色代表金属原子,蓝色代表轻元素(碳和氧),绿色代表背景;(c)四层金属配位的NiCo−UMOFNs的理论厚度[42];(d)二维层状CoNi(bdc)的TEM图像;(e)在O2−饱和的0.2 mol·L−1 PB电解液中的LSV测量(除空白CC外,样品的负载质量均为0.2 mg·cm−2)[43]Fig.5 (a)TEM image of NiCo−UMOFNs.The inset shows the Tyndall light scattering of NiCo−UMOFNs in an aqueous solution;(b)HAADF−STEM image of the(200)plane for NiCo−UMOFNs showing the hexagonal arrangement of the metal atoms.The pink colour represents metal atoms,blue is for light elements(carbon and oxygen),and green is for background;(c)Theoretical thickness of NiCo−UMOFNs with four metal coordination layers[42];(d)TEM images of 2D−CoNi−single−layer;(e)LSV measurements of 2D−Zn−single−layer,2D−Co−singlelayer,2D−Ni−single−layer,2D−CoNi−single−layer,and CC in O2−saturated 0.2 mol·L−1 PB electrolyte(all the loading mass of sample were 0.2 mg·cm−2 except blank CC)[43]
Li等[52]通过控制前体溶液中金属盐的比例制备了一系列不同比例的CoxFe−MOF 纳米片,如图7(a)、(b)所示,其中Co 或Fe 原子与NH2−BDC 部分配位导致表面上形成配位不饱和金属位点。FT−IR 结果表明纳米片层表面存在亲水羧酸基团,有助于增强Co3Fe−MOFs 表面对H2O 分子的吸附。铁原子的掺杂可以引起部分Co2+向Co3+的转化,从而发挥双金属之间的协同效应,有力地加速OER 反应。Xie 等[53]将Co、Fe双金属MOFs纳米片原位生长在NF上制备MIL−53(CoFe)/NF,如图7(c)所示。在1 mol·L−1KOH电解质溶液中,MIL−53(CoFe)/NF 达到100 mA·cm−2的过电位仅为262 mV,比单金属MIL−53(Co)/NF 和MIL−53(Fe)/NF 的过电势小,如图7(d)所示,说明铁离子的加入能够促使钴离子在MOFs中形成OER 活性位点,发挥双金属之间的协同作用从而促进电催化性能的提升。同时,在100 mA·cm−2的恒定电流密度下测试80 h,MIL−53(CoFe)/NF表现出卓越的稳定性,如图7(e)所示。
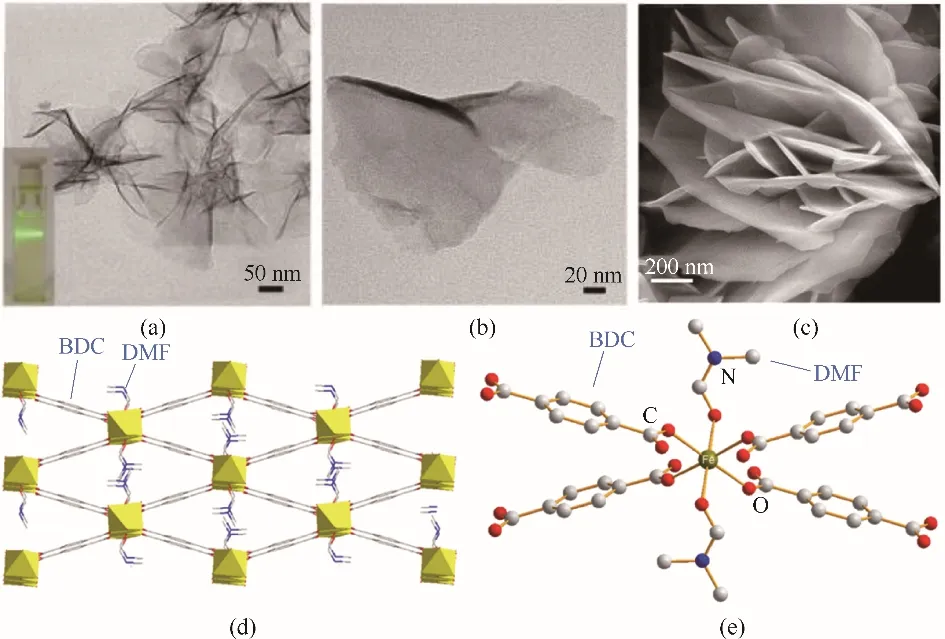
图6 (a),(b)Ni−Fe−MOF纳米片的TEM图像[45];(c)NiFe−UMNs的SEM图像[46];(d)Fe(BDC)(DMF)MOF的分子结构;(e)八面体Fe−O簇与有机连接基和溶剂分子的协同配位方式[48]Fig.6 (a),(b)TEM images of the Ni−Fe−MOF NSs[45];(c)SEM images of the NiFe−UMNs[46];(d)Molecular structure of Fe(BDC)(DMF)MOF;(e)Core octahedral Fe−O cluster with coordinated organic linker and solvent molecules[48]

图7 (a)Co3Fe−MOF的TEM图像;(b)Co3Fe−MOF的晶体结构及其与金属原子的配位方式[52];(c)MIL−53(CoFe)/NF的SEM图像;(d)在1 mol·L−1 KOH溶液中的LSV曲线;(e)在固定过电位为262 mV时,MIL−53(CoFe)/NF的电流密度曲线随时间的变化[53]Fig.7 (a)TEM image of the Co3Fe−MOF;(b)Crystal structure of Co3Fe−MOF and the corresponding coordination mode of metal atoms[52];(c)SEM image of the MIL−53(CoFe)/NF;(d)LSV curves in 1 mol·L−1 KOH;(e)Time dependence of the current density curve of MIL−53(CoFe)/NF at a fixed overpotential of 262 mV[53]
此外,在双金属二维MOFs 中引入另一种金属元素能够获得三元金属二维MOFs 材料。利用金属间的耦合作用能够大幅度降低OER 的过电位,进一步提高反应速率和效率。Qian等[55]利用金属醋酸盐和对苯二甲酸在室温合成具有泡沫结构的独特的镍基三元金属MOFs 纳米结构[(Ni2Co1)1−xFex−MOF−NF]。其在碱性条件下表现出优异的析氧反应活性,电 流 密 度 为10 mA·cm−2时,(Ni2Co1)0.925Fe0.075−MOF−NF 的过电位为257 mV。优异的OER 性能归因于Fe原子的掺杂提高了材料的电导率、引起Ni和Co 中心的部分电荷转移并激活了MOF 中三金属中心的协同效应。Ding 等[56]利用2−甲基咪唑作为配体与金属离子结合一步水热将2D MOF 纳米薄片原位生长在泡沫镍导电基底上,随后加入另一种金属离子形成三金属二维MOF 纳米片阵列(NiFeCo−ZIF/NF)。这种二维结构提供了更多的活性中心和更好的离子转移途径,Co2+和Fe3+的掺入与镍离子形成了三金属中心,能够调制电子结构从而大大提高材料的电导率。因此,三金属ZIF 纳米片表现出优异的OER 活性和稳定性,在1 mol·L−1KOH 中,100 mA·cm−2时的过电位为216 mV,Tafel 斜率为23.25 mV·dec−1。
1.2 配体调控
二维MOFs 材料中的有机配体能够有效调控金属中心的电子结构,优化MOFs 材料的本征活性[57]。同时改变有机配体与金属离子的比例能够实现MOFs纳米片的形貌与厚度调控[54],也可以利用新型的配体提高MOFs 纳米片的电导率[40,54],从而加速电子转移,提高其电催化活性。
Xue 等[57]报道了一种配体缺失策略来调节MOFs 的电子结构,使其具有更好的OER 性能。在MOFs 中,配体羧基二茂铁(Fc)部分替代了协调配位的BDC 配体,从而改变了金属中心的配位环境,将 新 形 成 的MOF 定 义 为CoBDC−Fc。CoBDC 和CoBDC−Fc 均具有纳米片形貌。利用傅里叶变换扩展X 射线吸收精细结构(EXAFS)光谱研究CoBDC和CoBDC−Fc 的Co 节点的局部配位,在CoBDC 中Co−O 的配位数为6.2,而在CoBDC−Fc 中Co−O 的配位数为4.4,说明在CoBDC−Fc中产生了不饱和Co位点。DFT 计算表明在CoBDC−Fc 中,金属中心Co2上的反应中间体的结合能得到了优化,因此具有优异的OER 活性。CoBDC−Fc−NF 只需要178 mV 的小过电位就可以获得10 mA·cm−2的电流密度,比CoBDC−NF(252 mV)和RuO2−NF(235 mV)小得多。因此,MOFs 纳米片的配体工程可以作为调控金属中心电子结构的一种有效方法,为潜在的电催化应用提供依据。
Jia 等[40]首次设计并合成了一种新型的酞菁镍基 二 维MOF(NiPc−MOF),其 组 成 成 分 是[Ni3(C32H16N16)]n。首先制备2、3、9、10、16、17、23、24−八氨基酞菁酸镍(NiPc−NH2),将制得的NiPc−NH2与镍(Ⅱ)盐(NiCl2·6H2O)反应,随后,NiPc−NH2单体相互连接形成了一个具有四重对称的巨大π−共轭2D MOF,如图3 所示。新型的酞菁镍基配体能提高MOF 材料的电导率(0.2 S·cm−1),有助于OER 过程中的电子传导。在电催化期间NiPc−MOF 中金属中心与配体中的氧原子之间形成的Ni−O 相互作用导致了微小的结构重排,这有利于产生OER 的反应中间体,从而提高其OER的活性。
Zhou 等[54]利用1,3,5−三(3,5−间二羧基苯基)苯(H6BHB)作为配体,Co 原子、Ni原子为金属中心,制备了一系列新型的结构一致的过渡金属MOFs[NH2(CH3)2] [M3(μ3−OH)(H2O)3(BHB)] (M3=Co3, Co2Ni,CoNi2, Ni3;分别命名为CTGU−10a1, b1, c1, d1)。每个BHB6−连接六个M3(μ3−OH)形成一个三维多孔结构,如图8(a)所示。通过改变金属与配体的比例会形成不同的形貌:如由纳米薄片组装成的分级纳米球CTGU−10b2(Co2NiMOF),如图8(b)所示,纳米片的厚度约为1.11 nm;由纳米薄片构建的分级纳米带CTGU−10c2(CoNi2MOF),如图8(c)所示,纳米片的厚度约为1.03 或1.05 nm。分级纳米结构由于配体与金属中心的相互作用使其本身具有增强的导电性,从而表现出优异的OER 催化活性和稳定性,如图8(d)所示。
1.3 复合结构调控
为了提升二维MOFs 的电催化性能,除了通过调节其组分以优化催化剂的本征活性外,还可以通过复合结构调控来增强反应动力学。特定的结构转变能抑制二维MOFs 的聚集,增大催化剂的活性表面积,暴露更多的活性位点,同时改善每个活性位点的可及性,进而提升催化剂的活性和稳定性。

图8 (a)CTGU−10a1的结构图(C:黑色,O:红色,Co:紫色);(b)CTGU−10b2的高分辨率TEM图像;(c)CTGU−10c2的高分辨率TEM图像;(d)在0.1 mol·L−1 KOH中,CTGU催化剂的LSV曲线[54]Fig.8 (a)Molecular structure of CTGU−10a1(C:black,O:red,Co:purple);(b)High−resolution TEM images of CTGU−10b2;(c)High−resolution TEM images of CTGU−10c2;(d)LSV curves of RuO2 and the CTGU electrocatalysts in the OER in 0.1 mol·L−1 KOH[54]
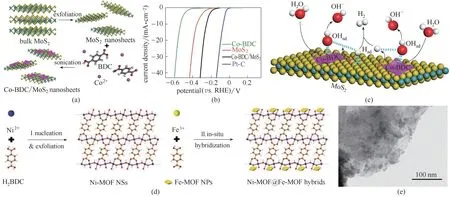
图9 (a)Co−BDC/MoS2杂化纳米片的合成过程示意图;(b)1.0 mol·L−1 KOH中扫描速率为5 mV·s−1时Co−BDC、MoS2、Co−BDC/MoS2、Pt−C的LSV曲线;(c)碱性条件下Co−BDC/MoS2杂化纳米片上的HER催化机理示意图[58];(d)Ni−MOF@Fe−MOF纳米片的合成示意图;(e)Ni−MOF@Fe−MOF的TEM图像[63]Fig.9 (a)Schematic of the synthesis process for Co−BDC/MoS2 hybrid nanosheets;(b)LSV curves of Co−BDC,MoS2,Co−BDC/MoS2,and Pt−C in 1.0 mol·L−1 KOH at a scan rate of 5 mV·s−1;(c)Schematic illustration of catalysis mechanism of alkaline HER on the Co−BDC/MoS2 hybrid nanosheets[58];(d)Schematic illustration of synthesis of Ni−MOF@Fe−MOF hybrid nanosheets;(e)TEM images of Ni−MOF@Fe−MOF hybrid[63]
通过有选择地将二维MOFs 与其他功能材料复合(MOF−MoS2[58],MOF−石墨烯[59],MOF−贵金属纳米粒 子[60−61],MOF−氧 化 物/氢 氧 化 物[62],MOF@MOF[63]等),可获得优化结构的基于MOFs 的杂化材料。二维MOFs 与其他材料复合产生的界面通常有利于提升电催化活性。Zhu 等[58]通过简便的超声辅助溶液法合成了一种新型的二维Co−BDC/MoS2杂化纳米复合材料,如图9(a)所示。研究表明MoS2纳米片显著抑制了Co−BDC 的聚集,并且所获得的Co−BDC/MoS2纳米片为催化反应提供了更大的活性表面积。羟基和H2O 分子能够在Co−BDC 纳米片表面发生有效的吸附和解离,从而为附近的MoS2提供足够的质子。XPS 结果表明,Co−BDC/MoS2间的相互作用能增大MoS2的电子云密度,减弱H*在MoS2表面的吸附强度,促进H*的重组和氢的释放,如图9(b)、(c)所示。因此,Co−BDC/MoS2具有优异的碱性HER 活性。在片层MOFs 结构中复合功能性纳米粒子是实现高效催化活性的有效策略。Xia 等[61]设计并合成了一种铂纳米颗粒(Pt NPs)嵌入超薄2D MOFs 纳米片的复合纳米材料,通过Pt 原子和MOFs 中金属中心之间的电子修饰来提高MOFs的OER 性能。与没有Pt 原子的材料相比,锚定Pt 的MOFs 纳米片可以削弱中间体在活性位点上的吸附强度,从而为该复合催化剂产生较低的能垒和较高的反应活性。Rui等[63]首次在室温下通过简单的逐步合成法制备了Fe−MOFs 纳米颗粒修饰的二维镍基MOFs 纳米片(Ni−MOF@Fe−MOF)作为碱性介质中的水氧化催化剂,如图9(d)、(e)所示。通过将电化学惰性的Fe−MOFs 纳米颗粒引入到活性2D Ni−MOFs 纳米片上,Fe−MOFs 颗粒能有效抑制Ni−MOFs 纳米片的堆积,从而促进固−液界面处更多活性位的暴露。同时,电化学活化后纳米片上形成介孔,有助于促进物质传递,极大提高了催化活性。异位透射电子显微镜和拉曼光谱结果揭示杂化MOFs在OER 期间被原位转化为氧化物纳米粒子,充当了真正的活性中心,而MOFs 中配体骨架的存在有助于抑制活性Ni−Fe氧化物纳米粒子的团聚。
在自支撑3D 导电基底上直接生长超薄二维MOFs,能抑制二维MOFs 纳米片的团聚,暴露更多的活性位点,提升MOFs 纳米片的导电性[44,47,64−66]。Duan 等[64]通过溶解−结晶机制在不同基底上原位生长金属−有机骨架的超薄纳米片阵列,如图10(a)所示。泡沫镍上的NiFe−MOF 纳米片阵列的独特分层多孔结构能够确保电极内物质的有效传输:泡沫镍的超大孔可促进电解质和气体产物的快速运输,而垂直排列的纳米片之间的开放孔、MOF 的微孔和介孔为催化反应提供了大量可接触的活性位点,有效缩短了离子扩散途径,如图10(b)、(c)所示。无黏结剂电极增强了催化剂与导电底物的接触,有助于有效的电子传输,从而提高催化活性。Wang 等[65]开发了一种在不同导电基底上将层状双氢氧化物(LDHs)原位转化成超薄双金属−MOFs 纳米片(BMNS)阵列的通用且简便的策略,如图10(d)、(e)所示。NiCo−BDC BMNSs 阵列具有快速的离子扩散速率,高效的电荷转移速率,高暴露的活性位点以及加速的气泡释放速率,比NiCo−BDC BMNSs 粉末和原始的NiCo−LDH NSs 阵列表现出更高的OER 活性和稳定性。这种原位合成的超薄MOFs 纳米片与导电底物结合紧密且牢固,有利于高效的电子转移,保证了电极结构的优异稳定性。
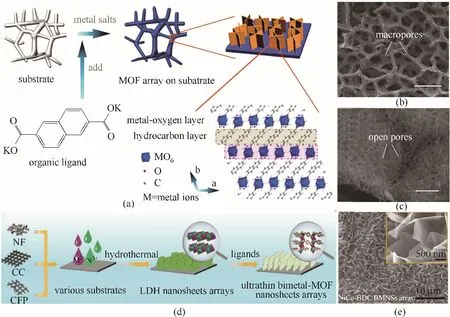
图10 (a)NiFe−MOF纳米片阵列的合成过程;(b),(c)NiFe−MOF的SEM图像[(b)的比例尺为300 mm,(c)的比例尺为1 mm][64];(d)在3D导电矩阵上制备超薄BMNS阵列的示意图;(e)NiCo−BDC BMNSs阵列的SEM图像(插图显示高倍放大SEM图像)[65]Fig.10 (a)Synthetic process of NiFe−MOF nanosheet array;(b),(c)SEM images[scale bars are 300 mm for(b)and 1 mm for(c)]ofNiFe−MOF[64];(d)Schematic illustration showing the fabrication of ultrathin BMNSs arrays on the 3D conductive matrix;(e)SEM images of NiCo−BDC BMNSs arrays(the inset shows the high magnifcation SEM images)[65]
2 二维MOFs 衍生物用于电催化水分解
以二维MOFs 为牺牲模板可以制备高效的电催化剂。在可控的温度和气氛下,二维MOFs 的金属中心能够转化为氧化物、磷化物、硫化物等化合物,而有机配体则转化为杂原子掺杂的碳材料。通过控制前体的组分和热处理方式,能够实现MOFs 衍生物组分及结构的调控,从而有效地提升催化性能。
2.1 氧化物
具有低成本、高活性、高热力学稳定性等优点的过渡金属氧化物是目前碱性介质中最具发展前景的电催化剂之一,但是较低的电导率限制了其在电催化领域的进一步应用。以二维MOFs 为前体能够制备负载过渡金属氧化物的二维导电碳材料,有效提高了催化剂的导电性和活性位点的分散性。通过引入氧空位缺陷[67−69]或构筑高价态的过渡金属活性位点[70−71]能有效提升二维MOFs 衍生氧化物催化剂的电化学性能。
Hu 等[67]在250℃的空气中煅烧超薄2D Ni 基MOF(2D Ni−MOF)前体,低温煅烧使MOF 的超薄二维形貌和多孔纳米结构保留,且纳米片中生成了高度分散的富缺陷的超小NiO 纳米颗粒(NPs),如图11(a)~(d)所示。二维纳米片中超小的NiO NPs 上存在的大量氧空位能够优化金属氧化物对OER 中间体的吸附能,且超薄二维纳米结构还有利于暴露大量的活性位点并加速电荷/物质传输。因此二维Ni−MOF−250 具有高的OER 活性和优异的稳定性,在410 mV 的小过电位下达到行业基准的1000 mA·cm−2电流密度,在110 mA·cm−2保持20 h电流密度几乎不变。
氧化物中不饱和金属活性位点和氧空位的存在能够有效提升材料的电化学性能[67−68]。氧化物中的不饱和金属位点通常作为催化反应的活性中心,具有较高的电催化本征活性[26,68],氧空位的存在则有利于优化金属氧化物对OER 中间体的吸附能,进而提高电催化性能[67,69]。Chen 等[68]通过使用O2−Ar 射频等离子体在碳布上合成具有不饱和金属位点和富氧空位的铁原子掺杂2D Co−MOF 多孔结构。随后,在高温下将具有丰富空位的Fe/Co−MOF 进一步碳化,获得了疏松且高度多孔的Fe1Co3/VO−800 纳米结构,如图11(e)~(h)所示。O2−Ar射频等离子体使得Co−MOF@CC 中的金属活性位点产生缺陷和大量的氧空位。Fe原子的掺杂有助于调整MOFs中两种金属的原子位点,使Co 活性中心与Fe 原子之间产生独特的协同效应,有效地调节催化剂的电子结构。所以,经过优化的Fe1Co3/VO−800 表现出出色的OER性能。

图11 (a),(b)二维Ni−MOF−250的SEM图像;(c),(d)2D Ni−MOF−250的TEM图像[67];(e)合成的Fe1Co3/VO−800作为高效的OER电催化剂的结构优势示意图;(f)O2−Ar射频等离子体对Co−MOF@CC在180 W改性后SEM图;Fe1Co3/VO−800的(g)SEM图和(h)TEM图[68]Fig.11 (a),(b)SEM images of 2D Ni−MOF−250;(c),(d)TEM images of 2D Ni−MOF−250[67];(e)Schematic for the synthesis process of porous Fe/Co−carbon with the microstructure inspired by the triangle shaped cheese;(f)Co−MOF@CC modified by O2−Ar RF plasma at 180 W;(g)SEM image and(h)TEM image of Fe1Co3/VO−800[68]
Li 等[69]通过简单的热解−氧化策略成功合成了由MOFs纳米片衍生的三金属Co2+/Fe2+/Ni2+碳纳米花电催化剂。不同的金属比例会影响最终催化剂的形貌和电化学性能。经过优化显示出Co0.2Fe0.8Ni−OCNF比双金属MOFs和其他金属比例的MOFs衍生的催化剂更优异的OER 活性和稳定性,在1.65 V 的电压下达到10 mA·cm−2的电流密度。三种金属在MOFs 中的均匀分布确保了衍生的N 原子掺杂碳材料中基于三金属/金属氧化物活性位点的高度分散,为催化反应提供了大量的活性位点。且不同金属组分之间具有丰富而强大的电子效应,可以诱导产生许多氧空位,从而有利于电催化性能的提升。

图12 中毒测试和用HCl蚀刻前后CoZn−NC−700的电催化性能(a)浸入O2饱和的0.1 mol·L−1 KOH之前(黑实线)和之后的ORR和OER LSV曲线,其中包括10×10−3 mol·L−1 KSCN(红色虚线)或10×10−3 mol·L−1 EDTA(蓝色虚线);(b)用0.1 mol·L−1 HCl蚀刻之前(黑色实线)和之后(红色虚线)的ORR和OER LSV曲线;(c)OER质量和比活性与Co3+/Co2+比之间的关系[70]Fig.12 The electrocatalytic performances of CoZn−NC−700 before and after poisoning tests and etching with HCl(a)ORR and OER LSV curves(at 1600 r·min−1)before(black solid lines)and after immersion in O2−saturated 0.1 mol·L−1 KOH that includes 10×10−3 mol·L−1 KSCN(red dashed lines)or 10×10−3 mol·L−1 EDTA(blue dashed lines)for 3 h;(b)ORR and OER LSV curves(at 1600 r·min−1)of before(black solid lines)and after(red dashed lines)etching with 0.1 mol·L−1 HCl;(c)Relationship between the OER mass and specifc activity and ratio of Co3+/Co2+[70]
研究表明高价态金属位点对OH−和OOH−的吸附是有利的,从而能促进OER 活性中间体OH*、O*和OOH*的形成[10,70]。Yin 等[70]通过热解含锌和钴的双金属有机框架制备了MO−Co@NC(M=Zn或Co)作为高活性OER 电催化剂,与基准的贵金属基催化剂相比,优化的CoZn−NC−700 显示出杰出的电催化活性和稳定性。图12(a)、(b)展示了催化剂CoZn−NC−700 中毒和HCl 刻蚀前后的LSV 曲线,可以观察到使用中毒剂和HCl 刻蚀后,CoZn−NC−700 的OER 活性大大降低,这表明催化剂的OER 活性主要由过渡金属基位点决定。通常高价态金属位点(如Co3+)对析氧反应中间体的吸附是有利的[10,70],图12(c)显示金属活性位点中Co3+/Co2+的比率越高,OER 质量和比活度越高。当催化剂的前体中含有Zn 元素时,Co3+/Co2+的比率会提高,从而有助于提高OER 活性。同时高温下Zn 元素的蒸发使得材料获得了高比表面积、高孔隙率和高电化学活性表面积,锌基和钴基物质之间的协同作用也有利于促进多壁碳纳米管(MWCNT)的生长,良好的MWCNT 和高度石墨化的NC 基质可以保证电催化过程中的电荷快速转移。Zhang 等[71]将超细CoFeOx纳米颗粒嵌入Co 基MOFs 的晶格中,再将其剥离成层状异质结构,形成含有无机纳米颗粒的单层非均质纳米薄片。HAADF−STEM 和XAS 分析表明,在金属氧化物纳米颗粒和单层MOF 基质的界面上形成了具有高价态的Co 位点,并改变了3d 电子构型,从而降低其对反应中间体的吸附能,增强了电催化OER性能。
催化剂的形貌和结构对电化学性能有很大影响。Zhou等[72]通过使用不同维度的金属有机框架为模板,制备了各种形貌的Co3O4/C 复合材料。其中2D MOFs 衍生的片状MOx/C 阵列具有独特的结构特征,揭示了其相对于1D 或3D MOFs 衍生物显著增强的催化活性和良好耐久性的本质。Guan 等[73]通过简单的碳化和氧化组合方法以二维钴基金属有机框架为前体制备了嵌入氮掺杂碳纳米壁阵列中的不规则空心Co3O4纳米球。这种由不规则中空氧化物纳米球和碳洋葱层形成的独特分层多孔结构包含许多高度应变的特殊位点,是优异的催化活性的来源。
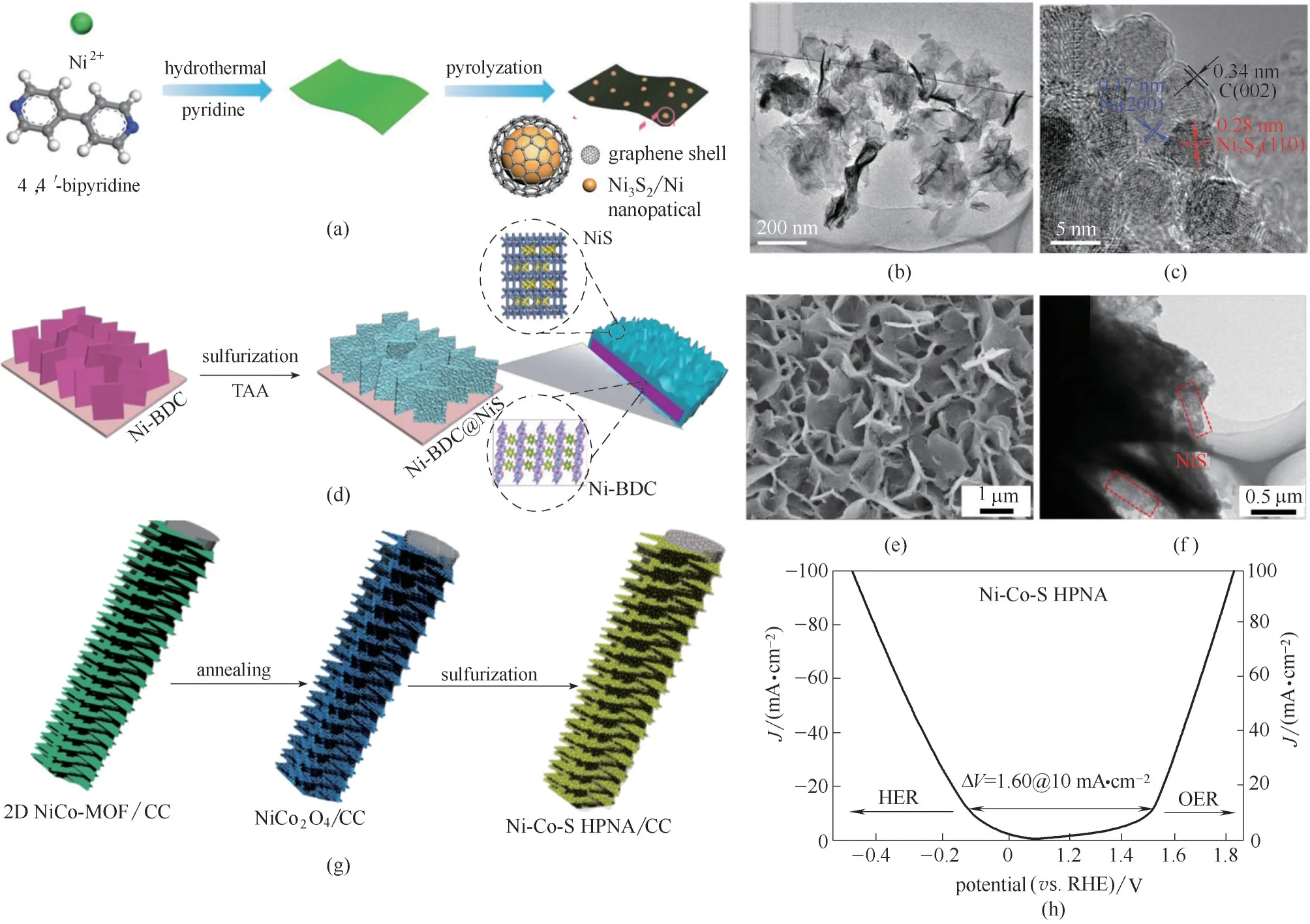
图13 (a)基于Ni−MOFs前体制备二维N原子掺杂Ni−Ni3S2@C纳米的策略示意图;(b)Py−1@SNC600的低放大TEM图像;(c)Py−1@SNC600具有清晰分辨晶格条纹的HRTEM图像[74];(d)Ni−BDC@NiS的合成策略图;(e)Ni−BDC@NiS的SEM图像;(f)Ni−BDC@NiS的TEM图像[75];(g)分级多孔Ni−Co−S HPNA/CC 纳米结构的形成原理图;(h)HER和OER在三电极电池中的Ni−Co−S HPNA极化曲线[78]Fig.13 (a)Schematic illustration of the preparation strategy of 2D N−doped Ni−Ni3S2@carbon nanoplates from Ni−based MOFs precursor;(b)Low magnifcation TEM image of Py−1@SNC600;(c)HRTEM image with clearly resolved lattice fringes of Py−1@SNC600[74];(d)Schematic illustrating for the fabrication of Ni−BDC@NiS array by in−situ growth of Ni−BDC and subsequent partial sulfurization for high performance of OER;(e)SEM image and(f)TEM image of Ni−BDC@NiS[75];(g)Schematic of the formation of the hierarchical porous Ni−Co−S HPNA/CC nanoarchitectures;(h)Polarization curves of Ni−Co−S HPNA for HER and OER in a three−electrode cell[78]
2.2 硫化物
近年来过渡金属硫化物(TMSs)被广泛应用于电催化水分解领域。电负性较强的S原子有利于调节金属中心的电子状态,优化反应中间体的吸附能,所以硫化物作为电催化剂具有良好的催化活性和快速的动力学特性。以二维MOFs 为前体衍生的TMSs 由于其高的导电性、充分暴露的活性位点、优异的催化性能在电催化析氢反应和析氧反应中被广泛研究。
Lin 等[74]首先利用吡啶(Py)作为抑制剂控制二维MOFs片层的厚度和宽度生成二维MOFs纳米片,之后自硫化将MOFs 转化为N 原子掺杂的Ni−Ni3S2@C 纳米片,如图13(a)~(c)所示,在Ni−Ni3S2纳米粒子周围均匀地包裹了一层超薄的无定形碳。在1 mol·L−1KOH 的电解质中,10 mA·cm−2时Ni−Ni3S2@C的OER 过电势为284.7 mV。可控的横向尺寸、二维形貌和多孔结构为催化剂提供了丰富的表面活性位点,增加了电解质的吸附和渗透通道。通过对热解时间和温度的控制,能够优化金属核中Ni与Ni3S2之间比例,使其协同调控材料的电子结构,从而实现更快的电荷转移。He 等[75]用硫代乙酰胺(TAA)将二维MOF 部分硫化,形成Ni−BDC@NiS 异质结构。其中,Ni−BDC 只有表面被部分硫化成NiS,内部依然为Ni−BDC,如图13(d)~(f)所示。部分硫化不仅可以保持MOF 丰富的多孔结构,而且由于形成的NiS 具有较高的电荷转移率和吸水能力,有利于促进OER反应的发生;同时,外部稳定的NiS可以作为内部MOFs的保护层从而使得OER 异质结构维持长时间稳定。
Co 基硫化物有许多存在形式,如Co9S8、CoS2等,均具有良好的电催化活性。Zhao 等[76]以Co 作为金属源,对苯二甲酸(H2BDC)和4,4′−(磺基双(4,1−苯基))二吡啶(SPDP)作为配体,形成一种新的Co−MOF,高温煅烧后,获得了N、O、S 杂原子掺杂的碳层包裹的Co9S8纳米粒子。Co9S8纳米粒子均匀地嵌在N、O、S 三元素掺杂的碳壳中,提高了电荷迁移能力,增强了材料的导电性,同时还能保护金属活性位点不被腐蚀,有利于提高OER 反应的活性和稳定性。在1 mol·L−1KOH 的电解质中,10 mA·cm−2时Co9S8@TDC−900 仅需较小的过电位330 mV,7 h 测试后电流密度保持初始值的95.5%,证明了Co9S8@TDC−900 对OER 的良好活性和稳定性。Wang 等[77]采用自下向上的方法在钛箔上生长二维的Co−MOF 纳米片,之后硫化生成N−CoS2@NC/Ti,CoS2纳 米 粒 子 被 包 裹 在 碳 层 中。在0.5 mol·L−1H2SO4溶液中,10 mA·cm−2时HER 过电位为105 mV,在1 mol·L−1KOH 中过电位为140 mV。CoS2@NC 中N 含量高,Co−Nx种类多,能够调节催化过程中间产物的电子结构,提供更多的活性位点,从而促进电化学催化反应动力学。
Chen 等[78]通过在Co−ZIF−L 纳米片中引入金属Ni 源,在碳布(CC)上生长了Ni 原子掺杂的钴基二维MOF 纳米片阵列,之后硫化形成了分级多孔Ni−Co−S 结构(Ni−Co−S HPNA/CC),如图13(g)所示。Ni−Co−S HPNA 杂化材料具有开放多孔的结构,有利于电催化过程中的物质交换和电子传递,加速反应动力学,增强电化学反应活性。基于其优异的HER 和OER 活性,使用催化剂作为双电极进行了整体水分解,如图13(h)所示,10 mA·cm−2时电池电压为1.62 V。且连续电解约24 h,其过电位和电流密度损失可忽略不计,表明电化学稳定性良好。双金属硫化物优异的电催化性能归因于Ni 原子的掺杂导致双金属原子之间产生协同催化作用,从而增加了催化剂活性位点的本征活性。
2.3 磷化物
过渡金属磷化物(TMPs)具有优异的导电性和类金属特性,是一类具有发展前景的电催化剂[79]。二维MOFs 磷化后形成磷化物纳米粒子均匀分散的二维碳层结构,包覆的碳层不但提高了材料的导电性,而且有效防止了大电流下磷化物颗粒的团聚以及催化活性表面积的下降,从而有利于催化剂稳定性的提高。
Zhai 等[80]将片状MOF(PPF−3)在高温下碳化形成Co@NC,随后磷化生成Co−P@NC,其中Co−P 由CoP 和Co2P 按照1∶1.03 的比例组成,退火温度为800℃时获得的催化剂的HER 和OER 性能最优。在N2饱和的0.5 mol·L−1H2SO4溶液中,Co−P@NC−800仅需98 mV 的OER 过电位即可达到10 mA·cm−2的电流密度,而在1 mol·L−1KOH 中,Co−P@NC−800 的OER 过电位为370 mV。超薄的二维MOF 纳米薄片转化成石墨碳薄层,使得催化剂具有较大的表面积和孔结构,暴露出更多可及的活性位点,有利于电子和物质的传递和反应。石墨碳中N原子的掺杂改变了碳材料固有电子结构,同时CoP 和Co2P 协同调制电子结构,从而有效地改善了材料的电催化性能。

图14 (a)CoP NS/CC合成工艺示意图;(b)CoP NS/CC的低倍和高倍SEM图像;(c)CoP NS/CC(+,−)和CC(+,−)在碱性和酸性|基底杂化水电解总效率的比较;(d)CoP NS/CC(+,−)在碱性和酸性|基底杂化水电解槽10 mA·cm−2恒流密度下的计时电位曲线[81];(e)在MOF合成初始阶段和24 h后的照片,标记为1、2、3的层分别对应于Co2+/Fe2+离子的溶液、隔离层和BDC溶液;(f)MOF装配过程示意图;(g)制备Co1−xFexP/C纳米薄片的示意图;(h)Co1−xFexP/C在1 mol·L−1 KOH中OER的LSV曲线[82]Fig.14 (a)Schematic diagram of the synthetic process of CoP NS/CC;(b)Low and high magnification SEM images of the CoP NS/CC;(c)Comparison of the alkaline and acid|base hybrid overall water−electrolysis efficiencies for CoP NS/CC (+,−)and carbon cloth(+,−);(d)The chronopotentiometric curve for CoP NS/CC(+,−)at a constant current density of 10 mA·cm−2 in the hybrid acid|base electrolyzer[81];(e)Photograph of a glass test tube at the initial stage of MOF synthesis and after 24 h.Layers labeled as 1,2,and 3 correspond to the solution of Co2+/Fe2+ions,the spacer layer and the BDC solution,respectively;(f)Schematic illustration of the MOF assembling process at layer 2;(g)Schematic illustration of the preparation of Co1−xFexP/C nanosheets;(h)LSV curves in 1 mol·L−1 KOH solution[82]
Zhu 等[81]在碳布上制备了ZIF−L−Co/CC 二维片层纳米阵列,之后将其氧化成Co3O4,再磷化成CoP NS/CC,如图14(a)、(b)所示。Co3O4相和纳米片阵列中磷的引入改善了电荷转移动力学,增加了催化活性位点,并促进了电解质的渗透和O2的扩散,有利于水分解。在酸性|基底双电解质水分解装置中(阴极和阳极CoP NS/CC 分别在0.5 mol·L−1H2SO4和1 mol·L−1KOH 中工作,预处理的萘酚膜作为电解分离膜),如图14(c)所示,10 mA·cm−2时仅需0.9 V的电压,比相应的单电解质碱性水解系统低0.77 V,且具有良好的循环稳定性,图14(d)中长期稳定性测试表明电池电压在10 mA·cm−2时每次补充酸性和碱性电解质会延长工作时间,电压可能回落至0.90~0.92 V。
通过在单金属磷化物中掺杂其他金属元素能够协同调控材料的电子结构,从而降低反应能垒,实现电催化性能的提升。Jiang 等[82]采用基于溶剂扩散调控的三层溶剂法合成了二维Co1−xFex−MOFs,磷化后形成Co1−xFexP/C,如图14(e)~(g)所示。通过XRD 和HRTEM 分析可知,Co1−xFexP/C 晶型与CoP 一致,说明Fe 元素掺杂成功。而且由于Fe 原子的掺杂,Co1−xFexP 纳米粒子的直径较CoP 减小,暴露出更多的活性位点。包裹的碳层有利于O*到Co0.7Fe0.3P纳米粒子的转移,使Co0.7Fe0.3P 纳米粒子的氧化更加完全,从而提高了其电化学性能。如图14(h)所示,在O2饱和的1 mol·L−1KOH 中,Co0.7Fe0.3P/C 达到10 mA·cm−2时的过电位为270 mV。Zhang 等[83]制备的多孔CoFeP TPAs/Ni 具有优异的稳定性,计时电流测试−20 mA·cm−2下的HER 和20 mA·cm−2下的OER性能持续超过100 h 其电流密度几乎没有发生变化。多孔CoFeP NPs/Ni 的晶体结构在长期的OER稳定性测试中变为非晶态。因此,多孔CoFeP TPAs/Ni 的高OER 性能可以归因于在OER 期间原位形成非晶态氢氧化物或氧化物。
Li 等[84]通过自下向上的方法,以Co 和M(Ni,Cu,Mn)为金属中心,二甲基咪唑为配体,合成了二维MOFs 纳米片组装的花状结构,之后将其磷化成双金属CoM−P−3DHFLMs,如图15(a)、(b)所示。在1 mol·L−1KOH 中,与Co−P−3DHFLM 相 比,双 金 属CoM−P−3DHFLMs 表现出更优的OER 性能,在10 mA·cm−2时CoNi−P−3DHFLMs 的 过 电 位 低 至270 mV,说明金属原子掺杂后能有效提高催化剂的OER 活性。Wu 等[85]在碳布上制备了ZIF−L−Co/CC二维片层纳米阵列,之后利用钼酸钠进行离子交换形成MoCo−LDH,再将其磷化形成Mo−CoP,如图15(c)、(d)所示。在1 mol·L−1KOH 溶液中,电流密度为−10 mA·cm−2时,Mo−CoP 的HER 过电势为40 mV。DFT 计算表明Mo 原子的掺杂导致ΔGH*值减小至接近零,从而大大增强了CoP 位点的HER 活性。在1 mol·L−1KOH 中,经过OER 活化后,Mo−CoP 原位转化 为Mo−CoOOH,10 mA·cm−2时,Mo−CoOOH 的OER 的过电势为305 mV。Mo 诱导的对H 的电子转移可以降低被吸附OH*中H—O 键的强度,从而促进潜在的脱质子过程,增强OER 活性。组成的全解水电解槽在低工作电压1.56 V下产生10 mA·cm−2的电流密度,在恒压1.7 V 下测试20 h,活性几乎没有损失,说明具有良好的稳定性。Zhou 等[86]采用离子交换法,将生长在泡沫镍上的ZIF−67纳米片阵列与Ni2+交换,形成NiCo(OH)x/NF,经过葡萄糖溶液的24 h 浸泡后,磷化得到产物Ni2P−Co2P@C/NF。Ni2P 的引入改变了Co2P 的电子结构,从而在Ni2P−Co2P 之间产生协同效应,极大地增强了Ni2P−Co2P@C/NF 的内在催化活性。
2.4 金属-碳复合物

图15 (a)CoM−P−3DHFLMs的形成过程示意图;(b)CoNi−P−3DHFLMs的SEM图[84];(c)中空Mo−CoP纳米阵列的制备方法;(d)中空Mo−CoP纳米阵列的SEM图[85]Fig.15 (a)Schematic illustration of the formation process of CoM−P−3DHFLMs;(b)SEM image of the CoNi−P−3DHFLMs[84];(c)Schematic fabrication process for hollow Mo−CoP nanoarrays;(d)SEM of hollow Mo−CoP nanoarrays[85]
二维MOFs 衍生的过渡金属−碳复合物通常是以M−Nx−C 和碳包覆的金属纳米颗粒(NPs)的形式存在,M−Nx−C 具有高的电催化活性,金属NPs 和碳载体协同发挥催化作用。通过引入第二种过渡金属元素形成双金属−碳复合物可以有效调节反应中间体的吸附能,进而降低反应能垒。
Xu等[87]使用二维Ni−MOF 作为前体在氮气气氛中高温退火制备了包裹几层氮掺杂石墨烯壳的Ni纳米颗粒(Ni@NC)用于电催化水分解,如图16(a)~(c)所示。Ni 核和氮掺杂的石墨壳之间能够协同优化石墨层的表面电子结构,从而调节反应中间体在石墨表面的吸附能,增强电催化活性。退火温度对Ni NPs 周围石墨层的含量、厚度和结构组成有很大影响。通过优化退火温度,获得含适量石墨层的Ni@NC−800(800 为温度),该催化剂在碱性溶液中表现出优异的HER和OER催化活性和耐久性。
为了更好地调节MOFs 衍生的过渡金属纳米粒子的分散程度,Wang 等[88]通过在钴基金属有机框架中引入易挥发的锌元素形成叶状双金属(Co/Zn)沸石咪唑酸盐骨架(ZIF−L)并热解,高温下锌原子挥发,生成了封装在氮掺杂碳纳米管中的高度分散的Co纳米颗粒(Co−N−CNT),如图16(d)~(f)所示。优化的Co−N−CNT 显示出出色的OER 催化活性和稳定性,高度分散的钴和氮原子配位形成的Co−N−C 结构是优异的OER 活性中心。CNT 封装的钴纳米粒子增加了碳化过程中表面碳层的石墨化程度,同时氮在碳晶格中的掺杂有效促进了电子转移,且碳壳的形成不但阻止了Co 纳米颗粒在催化过程中的氧化和聚集,而且增强了电子转移到表面的能力。Sun等[89]通过在H2/N2中热解镍泡沫上片状镍基MOF前体获得一种封装非掺杂碳基体中的镍纳米粒子,如图16(g)~(i)所示。碳化过程中生成的碳纳米管末端的超小Ni NPs 是高活性的催化位点,同时,在石墨碳基质中均匀分散的Ni NPs 能够通过调节石墨层的局部电子结构来优化反应中间体的结合能,从而实现高效催化。此外,具有较大表面积的MOF 层状纳米片衍生的碳层以及生成的碳纳米管极大缩短了电子转移路径,促进了电荷和物质传输,也有利于电解质的渗透,加快了反应动力学。
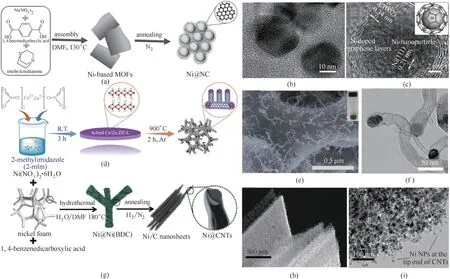
图16 (a)基于Ni的MOF前体衍生制备Ni@NC材料的过程示意图;(b)Ni@NC−800的TEM图像;(c)Ni@NC−800的HRTEM图像,插图是Ni@NC的结构示意图[87];(d)Co−N−CNT合成过程的示意图;(e)Co/Zn(1:1)杂化ZIF−L和(f)其生成的Co−N−CNT的SEM图像,插图显示了制备好的粉末的相应照片[88];(g)NF@Ni/C−600的合成过程的示意图;(h)NF@Ni/C−600的SEM图像;(i)NF@Ni/C−600的TEM图像[89]Fig.16 (a)Schematic illustration of the preparation process of Ni@NC materials from Ni−based MOF precursors;(b)TEM images and(c)HRTEM images of the Ni@NC−800 sample.The inset of(c)is a schematic illustration of the Ni@NC structure[87];(d)Schematic illustration toward the synthetic process of Co−N−CNTs;SEM images of(e)Co/Zn(1∶1)hybrid ZIF−L and(f)its resultant Co−N−CNTs.The inset images show the corresponding photographs of as−prepared powders[88];(g)Schematic illustration of the preparation process of NF@Ni/C composites from the NF@Ni(BDC)precursor;(h)SEM and(i)TEM images of NF@Ni/C−600[89]
Huo等[90]通过热解2D片状Cu(OH)2/ZIF−L 材料,制备了封装CuCo合金纳米颗粒的氮掺杂碳纳米叶。首先,通过超声诱导Cu(OH)2纳米线自组装合成叶状Cu(OH)2,然后将预先制备的Cu(OH)2、Co(NO3)2和2−甲基咪唑混合形成叶状Cu(OH)2@ZIF−L,最后在氩气气氛中900℃下热解获得CuCo@NC 材料。ZIF−L−Co 前 体 中Cu(OH)2的 掺 杂 量 会 影 响 最 终CuCo@NC 材料中Cu 和Co 位点的相应状态和缺陷结构,且引入Cu 原子改善了材料的电导率,而且还在电催化剂中生成更多的活性位点和孔隙率,有利于促进传质和电子转移并改善催化作用。同时,氮原子的掺杂尤其是石墨氮和吡啶氮为催化析氧反应提供了更多的活性位点。碳层中Co 原子与N 原子之间的配位能够调节析氧中间体的吸附速率,有助于提高CuCo@NC 催化剂的OER 电催化活性和稳定性。
2.5 其他衍生物
除了前文介绍的氧化物、硫化物、磷化物、金属−碳复合物外,二维MOFs 的其他衍生物如氮化物、氟化物、硒化物等也被应用于电催化水分解领域。
Xu 等[91]将碳布上MOF 前体转化的共掺杂Fe−Co4N@N−C 纳米片阵列作为电催化剂,由于金属中心与吡啶氮的强配位效应,在OER 过程中,富集的Co3+位点促进了目标中间产物*OOH 的形成,同时降低了电荷转移阻力。因此,这种电催化剂对OER 表现出很高的催化活性。Guan 等[92]通过将二维层状结构的Co−ZIF−L 碳化和氮化,得到Co/CoNx纳米粒子嵌入的NC 纳米阵列。N 掺杂碳纳米阵列与柔性基底具有直接的电/机械连接,有利于电子的传递和结构稳定性的保持。由Co、Co2N 和Co4N 组成的混合非均相纳米颗粒能够为催化OER 反应提供丰富的活性位点、高密度的界面和缩短的离子扩散路径,并确保了整个电极的高电导率,从而增强了材料的催化性能。
氟化物是一种新型的电催化剂,元素氟电负性高,能够在催化剂中调节金属的电子状态,从而提升其电催化活性。Rodenas 等[93]通过组装和热解复合前体构成的二维钴基MOFs 晶体和石墨薄片制备了CoF2/碳电催化剂,利用有机配体中的氟形成纳米尺寸的CoF2晶体,而且多余的氟嵌入在MOFs 衍生的非晶碳基体中。掺杂的氟原子通过迁移使碳载体功能化,这一作用能够产生积极的电催化效应,如修复氧化位点从而增强抗腐蚀稳定性;增加疏水性从而有助于从电催化剂表面有效地释放O2气泡;提高导电性从而有效地提高电子转移速率。最终在碱性OER 过程中,CoF2/碳催化剂表现出优异的电催化活性。

图17 (a)MOF−Co纳米片形成多孔CoSe2和CoSe2的示意图;(b)~(d)在不同放大倍数多孔CoSe2的SEM图[95];(e)CFC上掺杂锌原子的CoSe2的合成方法示意图;(f),(g)在不同放大倍数的CFC上生长的掺杂锌原子的CoSe2纳米片[96]Fig.17 (a)Schematic illustration of the formation for porous CoSe2 and CoSe2 from MOF−Co nanowall;(b)—(d)SEM images of porous CoSe2 at different magnification[95];(e)Schematic illustration of the synthesis procedures of Zn−doped CoSe2 on CFC;(f),(g)Zn−doped CoSe2 nanosheets grown on CFC at different magnification[96]
硒化物近年来发展较快,被逐渐应用于电催化领域。带负电的硒位点和金属与硒带中心之间的3d−2p 排斥力能够更快地输送双氧中间体,增强反应动力学[94]。Chen 等[95]合成了多孔CoSe2纳米阵列,如图17(a)~(d)所示。XRD 表明多孔CoSe2的结晶度比CoSe2的结晶度好,是良好的电子转移载体,具有较好的离子扩散和电荷转移性能。经过刻蚀和退火后,吡啶N 和吡咯N 仍然保留于多孔CoSe2中,增加了活性位点。同时,多孔结构暴露出的丰富活性位点以及自支撑结构固有的优异导电性,共同确保了多孔CoSe2的出色OER 性能,电流密度为10 mA·cm−2时 仅 需1.52 V 的 低 电 位。Dong 等[96]以 二 维MOFs 为前体,经硒化反应合成生长在碳布上的Zn原子掺杂的CoSe2纳米薄片,如图17(e)~(g)所示。在1 mol·L−1KOH 中,电流密度为10 mA·cm−2时,CoSe2/CFC 的过电位为356 mV,且稳定性良好。XPS 结果表明锌原子成功掺杂到CoSe2晶格中,锌原子的掺杂引起电子结构的改变及缺陷/活性位点的增加,对于OER活性起到重要的促进作用。
Wu 等[97]制备了以二维氮掺杂碳(NC)为载体的Co−Fe−P−Se 纳米粒子,构建了自支撑三维纳米结构。二维纳米片暴露出大量的活性位点,能促进物质传递。纳米片组装的三维结构有利于电解质的浸润,为反应产物的扩散提供了丰富的通道。此外,碳基体中的氮掺杂以及Co 和Fe 的共存都对催化性能的提升有显著作用。硒化物和磷化物的共存有效提高了OER催化性能。1 mol·L−1KOH中,电流密度为10 mA·cm−2时,Co−Fe−P−Se/NC 的过电位仅为270 mV,比Co−Fe−Se/NC(300 mV)和Co−Fe−P/NC(289 mV)都低,说明同时掺杂P、Se 元素能有效增强OER活性。
2.6 结构调控
结构调控对提升二维MOFs 衍生物的电催化性能至关重要,通过构筑中空/多孔结构、自支撑结构能够有效提高活性表面积、有利于传质。此外,还可以进一步制备单原子结构来提高活性位点的密度。研究表明,在催化剂表面构造空位/缺陷能够有效调节催化剂的电子结构,降低催化过电势,提升活性和稳定性[67,98−99]。
纳米材料的表面结构对实现高效、稳定的电催化至关重要。Hu 等[67]通过控制热解温度,将超薄二维Ni−MOF 在低温下热解形成缺陷丰富的超小NiO纳米粒子,小尺寸的金属氧化物容易暴露更多的表面缺陷(即氧空位),如图11(a)~(d)所示,通过XPS 和电子自旋共振(ESR)证明氧空位的存在,丰富的氧空位能优化材料对于反应中间体的吸附能,使其具有优异的OER 活性。Chen 等[68]利用氧等离子体处理ZIF−L 纳 米 片,如 图11(e)~(h)所 示,O2−Ar 射 频(RF)等离子体确保了两个关键的效应,即氧空位的产生以及不饱和活性金属氧化物位点在骨架中的形成和修饰,从而导致较高的OER 性能。Zou 等[100]采用硼氢化钠将二维MOF 还原成具有大量缺陷(氧空位、晶界)的无定形的核壳结构r−CoFe。在HRTEM 图像中清晰显示出高度混乱和破碎的条纹,如图18(a)所示,杂乱晶界处的大量缺陷能够暴露更多的活性位点,XPS 也证明了大量氧空位的存在。由于缺陷的存在,r−CoFe 的带隙能量(Eg)值大大降低,表现出较高的导电性和快速的电子跃迁,从而使其具有优异的OER活性和稳定性。
由于MOFs 晶体具有金属中心高度有序且均匀分布的特点,被认为是制备高密度M−Nx−C 活性位点的单原子电催化剂(SACs)的理想前体。二维MOFs 衍生的SACs 继承了MOFs 前体的许多优点,如高的孔隙率和大的表面积,有利于物质扩散和活性位点的充分暴露,在电催化应用中表现出优异的催化性能[101]。Zang等[102]通过高温将二维片层ZIF−L热解后,用酸将多余的金属Co 团簇去除,制成了包裹在氮掺杂的碳中的单原子Co。分散良好的Co 单原子通过N—Co 键与碳网络连接,同时,Co 金属团簇的去除产生了额外的孔隙和活性表面积。与含有过量Co 纳米颗粒的电催化剂相比,单原子Co 原子利用率高,能够暴露更多的活性位点,具有独特的催化性能,表现出更低的OER过电位。

图18 (a)r−CoFe的HRTEM图[100];(b),(c)大孔CoFeP TPAs/Ni的SEM图[83]Fig.18 (a)HRTEM image of r−CoFe[100];(b),(c)SEM image of macroporous CoFeP TPAs/Ni[83]
低密度、高渗透的空心结构有利于调节反应物的局部微环境,为快速的物质扩散提供大量通道。微孔(<2 nm)和中孔(2~50 nm)是扩大比活性表面积的关键,而大孔(>50 nm)则为反应物提供了充足的开放通道,使反应物可以同时接触内部和外部活性位点[35]。由于MOFs 具有开放结构和孔径可调的优点,因此可以通过构建更复杂的空心和多孔结构来有效改善电催化剂的物理和化学性能。Wu 等[85]利用钼酸铵将ZIF−L 纳米片刻蚀成中空结构,如图15(c)、(d)所示,Kong 等[103]利用Zn2+将ZIF−L 刻蚀成中空结构,经过后处理后虽然形成的产物化学结构不同,但是中空结构缩短了离子扩散长度,使材料具有更高的比表面积,为催化反应提供了充足的反应位点,从而提升了最终的电催化性能。Chen 等[95]利用乙醇和水的刻蚀作用,将ZIF−L 刻蚀成多孔的CoSe2结构,如图17(a)~(d)所示,多孔结构增大了比表面积,使活性位点与电解液充分接触,促进催化反应的发生。纳米孔不仅可以提供丰富的反应位点和较短的离子扩散距离,而且可以在电化学反应过程中释放应变,使产生的气体可以很好地被释放。Zhang 等[83]利用NaOH 对CoFe−MOF 进行刻蚀,之后将其磷化,生成多孔的CoFeP TPAs/Ni,如图18(b)、(c)所示。通过刻蚀在平面上形成的大的连通孔隙可以增加活性位点,提高有效比表面积。同时有助于促进电解质的输运和离子的扩散,加快反应动力学。
自支撑结构是将二维MOFs 纳米片垂直生长在导电基底上,如碳布(CC)[78,81,85,95−96]、金属导电 基底[77,89−90]等,通过后续处理将其转化成各种MOFs 衍生的二维纳米片阵列。自支撑结构有许多优势:(1)二维材料与导电基底结合能够防止片层之间团聚,暴露出更多的活性位点,提高催化效率;(2)纳米阵列内部的开放空间可以促进电解液的渗透,增加电解液与活性位点的接触,有利于提高电催化性能;(3)活性材料与导电基底之间直接的电子和机械连接有效地保证了电子转移路径,避免了使用额外的黏合剂,提高了材料的导电性,确保了催化过程中电子的快速传输;(4)开放的空间有利于产生的气体快速释放,加快物质传输,提高催化反应动力学;(5)导电基底的机械性能优异,能够承受大电流,有利于改善反应的长时间稳定性。许多研究工作将MOFs 纳米片原位生长于导电基底上[78,81,96],避免了黏合剂的使用,有效增强了材料的导电性。纳米片垂直生长,片与片之间形成开放的大孔结构,如图13(e),14(b),17(b)所示,有利于电解液的渗透和气体的释放。由于导电基底机械性能优异,能够承受长时间大电流工作,Zhang等[83]在泡沫镍基底上制备了二维MOFs 衍生的多孔CoFeP TPAs/Ni 用于电催化水分解。在1 mol·L−1KOH 中,电流密度为20 mA·cm−2下持续电解超过100 h,催化剂活性几乎没有损失,表现出优异的稳定性。多孔CoFeP TPAs/Ni相对于其他样品Rct较小,在HER 和OER 过程中电极与电解质之间的电荷转移更快。
3 总结与展望
二维MOFs 及其衍生物具有结构独特、比表面积大、可裁剪性强等优点,是近期电催化分解水领域的研究热点。MOFs 的结构多种多样,组成丰富,通过选择金属中心和设计官能化配体能实现其电催化性能的优化。此外,以二维MOFs 为前体,经高温热解可得到多种衍生物。通过选用不同结构的二维MOFs 作为前体,能够实现衍生物组成和结构的调控,从而确保其电催化水分解性能的提升。基于此,本文介绍了近年来二维MOFs 及其衍生物在电催化分解水领域的研究进展,尽管如此,未来二维MOFs 及其衍生物作为催化材料仍面临许多的问题与挑战。
(1)尽管MOFs 材料丰富多样,但目前大多数工作报道的二维MOFs 还是一些常用的ZIF、PBA 和MIL 系列,不利于丰富此类材料的结构并限制其催化性能的提升。在未来仍需发展新型结构的二维MOFs 以及相应的MOFs 衍生材料,丰富其组成及结构,以期得到性能更为优异的催化材料。
(2)二维MOFs 作为电催化剂表现出良好的电化学性能,但是其导电性普遍较低。未来可以开发导电二维MOFs,从而保证电子的快速传输。还可以通过调控反应条件,优化二维MOFs 衍生碳材料中的碳缺陷位结构以及石墨化程度,提高其电导率。此外,可以进一步与导电性好的材料(如石墨烯,碳纳米管等)复合制备电催化剂,提高导电性和催化活性。
(3)制备具有纳米形貌和结构更复杂的二维MOFs 或二维MOFs 衍生物可实现活性位点的有效分散,从而增加活性位点数量。此外还可以通过优化二维MOFs 的衍生条件来避免高温过程中片层材料的聚集和孔结构的坍塌,避免活性位点的损失,提高催化活性和稳定性。
(4)此外,从电催化的实用角度出发,设计稳定性优异的、能适应广泛pH 范围的二维MOFs 基催化剂将更具吸引力。目前,二维MOFs 及其衍生物材料作为电催化剂时稳定性较差,容易溶解在电解液中造成催化剂的损失。可考虑在二维MOFs 表面包覆碳壳层或者与其他稳定性较好的材料复合来提高稳定性。此外,目前大多数的二维MOFs 及其衍生物只能在碱性条件下使用,在酸性环境下的稳定性普遍较差,所以迫切需要发展广泛pH 范围下具有高活性和稳定性的电催化剂。
总之,基于二维MOFs 及其衍生物的催化材料具有比表面积大、孔隙率高、结构可控等优势,在电催化分解水领域显示出优异的性能。但目前上述进展还限于实验室研究阶段,研究人员需要进一步开展研究来寻找低成本、高制备效率和稳定性强的二维MOFs 及其衍生物材料,以推动该类材料在电催化分解水领域的实际应用。
