过渡金属氧化物催化析氧反应研究进展
2020-09-29张伶陈红梅魏子栋
张伶,陈红梅,魏子栋
(重庆大学化学化工学院,洁净能源与资源利用化工过程重庆市重点实验室,重庆400044)
引 言
自第一次工业革命以来,化石能源被过度开采与使用,造成的能源危机与环境问题,为人类社会可持续发展带来了巨大挑战,开发清洁的、可再生的替代能源是亟待解决的全球性问题[1−2]。氢能来源广泛、能量密度高、无污染和零碳排放,被认为是21 世纪最具发展潜力的清洁能源。在多种氢气制备方法中,立足于未来碳中性甚至负碳的电解水制氢,尤其是利用可再生能源的电解水制氢技术的工业化应用对推动氢能经济的发展具有重要的科学和社会意义[3−5]。
电解水制氢过程包含阳极析氧反应(OER)和阴极析氢反应(HER)[6−7]。水分子中氢氧键能高达498.7 kJ/mol,电解水制氢过程需要高活性的析氢和析氧电催化剂,以提高反应速率,降低过电位,增加能量转换效率。经过不懈的努力,研究者们在析氢催化剂开发上取得了一些成就,成功制备了一系列具有较高活性和稳定性的析氢催化剂,其中以过渡金属磷化物最为突出[8]。比如,Liu等[9−11]以过渡金属(M= Fe,Zn 等)掺杂的CoP 用作高性能析氢电催化剂,Chen 等[12]以Mn 掺杂的CoP 用作析氢催化剂。然而,阳极析氧反应涉及复杂的4电子转移过程,过电位往往高达数百毫伏,成为电解水制氢过程的速率控制步骤。因而,高性能析氧催化剂对电解水制氢过程尤为重要[13−14]。贵金属基析氧催化剂(如RuO2和IrO)活性高,但价格昂贵,稳定性不理想[14]。因此,开发兼具高活性和高稳定性的非贵金属基析氧催化剂,是实现电解水制氢技术工业化普遍应用的关键。
在众多的析氧催化剂中,晶体结构多样、储量丰富、制备容易、环境友好、析氧活性较高的过渡金属氧化物(TMOs),被视为析氧反应的理想电催化剂,受到了研究者们广泛关注。过渡金属氧化物常见的晶体结构有钙钛矿型[15−25]、烧绿石型[26]、层状双氢 氧 化 物[27−31]、岩 盐 型[32]、金 红 石 型[33−34]、水 钠 锰 矿型[35−38]、尖晶石型[16,39−49]等。过渡金属氧化物在制备过程中往往不需要保证无水无氧等苛刻的反应条件,且不会产生对环境有害的气体或固体。虽然已有文献对石墨烯负载的过渡金属氧化物用作析氧催化剂的研究进行了系统总结[50],然而在优化过渡金属氧化物析氧催化剂活性和稳定性策略方面,还缺乏系统全面的总结。为进一步推动析氧催化剂非贵金属化研究,加速电解水制氢技术的工业化进程,本文将总结分析近年来在提升和增强过渡金属氧化物析氧活性和稳定性方面的代表性研究工作(图1),并对其未来的发展提出建议与展望。
1 过渡金属氧化物析氧活性增强研究
1.1 电催化析氧机理
电解水制氢析氧过程的总反应因电解液酸碱性不同而存在差异[51]。
碱性电解液中:

酸性电解液中:

无论电解液呈酸性还是碱性,析氧反应都包含复杂的4 电子转移过程,涉及3 种吸附物种(*OH,*O,*OOH),但具体反应机理仍受电解液酸碱性影响[52−54]。

图1 过渡金属氧化物析氧催化剂的优化策略Fig.1 Strategies for optimizing the OER performance of transition metal oxides (TMOs)
碱性电解液中:

酸性电解液中:

在反应过程中,反应活性中间物种在催化剂表面吸附,反应速率与反应中间活性物种的吸附自由能直接相关[52,55]。另外,晶格氧过程,即来自催化剂晶格中的氧参与析氧反应,对析氧反应速率提升也有很大的促进作用[55](图2)。
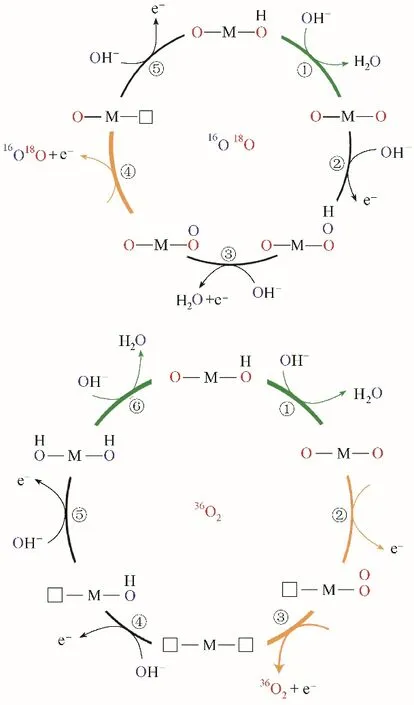
图2 两种可能的晶格氧参与析氧反应的机理[55]Fig.2 Two possible lattice oxygen involved OER mechanisms [55]
1.2 析氧催化剂电化学性能指标
析氧催化剂电化学活性的主要评价指标有过电位(ŋ)、Tafel 斜率(b)、交换电流密度(j0)、电化学活性面积(A)以及稳定性[56]等。
在一定的电流密度下,过电位越低,析氧电催化剂的表观活性越高。几何电流密度为10 mA·cm−2时的过电位,是用来表征析氧电催化剂活性的重要指标之一[57−58],几何电流密度为100 mA·cm−2和500 mA·cm−2时的过电位,则是评价析氧电催化剂工业化前景的重要指标[58]。通过线性伏安扫描法(LSV)得到的极化曲线,能快捷地获得析氧催化剂在不同电流密度下的过电位[59]。对线性伏安扫描法(LSV)得到的极化曲线进行数学运算,可以得到Tafel斜率(b)与交换电流密度(j0)[56]。Tafel斜率表征的是电流密度随过电位的增加速度,较小的Tafel斜率意味催化剂拥有更快的电极反应动力学。电极处于平衡状态时,阴极反应和阳极反应的电流密度相等,对应的电流密度就是该电极的交换电流密度(j0)。交换电流密度是一个热力学概念,反映了析氧电催化剂的本征活性[59]。
电化学活性面积指的是具有电化学反应活性的电极面积[60]。电化学反应是异相反应,由于催化剂表面存在许多细小的突起、沟壑等,催化剂的电化学活性面积往往比电极的表观面积大[60]。一般来讲,电化学活性面积越大,暴露的活性位点就越多,催化剂的电化学活性也就越高。然而,很多时候需要比较催化剂的本征活性,因此,准确确定电化学活性面积对于电化学析氧反应而言至关重要。常用的测定电化学活性面积的方法有三种:(1)利用欠电位沉积法测定析氧电化学活性面积[61−62];(2)利用氧化物还原峰测定电化学活性面积[63−64];(3)利用循环伏安法测定电化学活性面积[57−58,65]。
稳定性是析氧电催化剂的关键指标之一。恒电流/恒电压极化常用于考察催化剂的稳定性。恒电流/恒电压极化是指在电解池上施加一个恒定电流/电压,记录工作电极上电位与时间(P-t)或电流与时间(i-t)的变化曲线。通过恒电流/恒电压极化,可以考察催化剂在长时间电解过程中电流与电位的变化,进而判断催化剂的稳定性。常用的恒定电流一般为10 mA·cm−2或者20 mA·cm−2。在工业应用中,催化剂往往是处于较大的电流密度下,因此,在更大的电流密度下(往往达到300 mA·cm−2)测定催化剂的稳定性是很有必要的[66]。
1.3 析氧活性增强策略
计算化学的快速发展,使水电解制氢析氧催化反应机理在分子−原子水平上的研究得以实现。根据Sabatier 原理,催化活性中心应具有与活性中间物种适当的结合能,理想的催化剂表面对活性物种的吸附既不能太强也不能太弱[67]。由于析氧过程涉及3 种含氧活性中间物种(*OH,*O,*OOH)的吸附,使过渡金属氧化物表面析氧活性中间物种吸附自由能的优化变得复杂而困难[21,68]。对于析氧反应,有研究者提出以*O 和*OH 的吸附Gibbs 自由能变之差(ΔGO−ΔGOH)作为催化剂活性评价指标[55],鉴于该过程的复杂性,研究者们更倾向于利用理论过电位评价催化剂的析氧活性。近年来,研究者在过渡金属氧化物析氧催化剂方面做了大量工作,催化剂的析氧活性得到了很大提高。下面将从杂原子掺杂、异相界面构筑、缺陷位调控、结构无定形化和形貌调控等角度,分别阐述过渡金属氧化物析氧活性增强策略的研究进展。
1.3.1 杂原子掺杂 杂原子掺杂易于实现,因而成为最常用的催化剂活性调控手段。通过精细调控杂原子掺杂,能够优化反应活性中间物种在过渡金属氧化物表面的吸附自由能,提升析氧催化活性。将Ce掺入以Au为载体的NiOx中[69](表示为Ce−NiOx/Au),当Ni和Ce的原子比为95∶5时,Ce位点和Ni位点的析氧计算理论过电位分别为440 mV 和370 mV[图3(a)、(b)],远小于NiOOH 晶体中(0112)面上的桥式位点和顶式位点的计算理论过电位(分别为670 mV 和1020 mV)[图3(c)]。以Ni、Fe、Co 三种元素形成的复合氧化物[70],在Ce 掺杂后析氧活性得到了很大提高[图3(d)、(e)],特别是Ce掺杂比例较高的样品(Ni0.3Fe0.07Co0.2Ce0.43Ox),由于在析氧过程中原位形成了大小为3˜5 nm的萤石型CeO2和多金属的岩盐型氧化物结构,析氧活性更加优异[32]。
除了Ce,掺杂Fe[74−75]、Mn[72,76]、V[71,77−82]、Cr[83−84]和Ti[85]等原子对增强过渡金属氧化物析氧活性都行之有效。将Ti 掺杂到超薄NiO 纳米片[85],形成直径和厚度分别为约4.0 nm 和约1.1 nm 的双金属NiTi 氧化物超薄纳米片,在析氧电流密度为10 mA·cm−2时,过电位为320 mV。TiO2对双金属NiTi 氧化物中NiO 的高指数晶面起到了稳定作用,使更多未配位的Ni3+原子得到暴露,析氧反应活性得以显著提高[85]。将V 掺入CoO 中[71,77,79−80],得到Co、V 双金属氧化物a−CoVOx,当析氧电流密度为10 mA·cm−2时,过电位仅为347 mV[图3(f)]。理论计算表明,V掺杂优化了Co位点对OH 的吸附强度,增强了析氧活性[图3(g)]。此外,在无定形Co、V 双金属氧化物中掺杂V[80],也得到类似的结果。掺杂Mn 的CoMn 水合碳酸盐(CoMn CH)[72],当析氧电流密度为30 mA·cm−2和1000 mA·cm−2时,过电位仅分别为294 mV 和462 mV [图3(h)]。将电化学惰性材料Al 掺入Co3O4中[73][图3(i)、(j)],得到的样品Co1.75Al1.25O4在析氧电流密度为10 mA·cm−2时,过电位降低至248 mV [图3(k)]。然而,酸性介质中的非贵金属电催化析氧反应则是一个非常前沿的研究领域。Huynh 等[86]和Moreno−Hernandez 等[87]已分别研究了Fe 掺杂的CoFePbOx和Ni 掺杂的NixMn1−xSb1.6−1.8Oy在酸性介质中用作稳定、高活性的析氧催化剂。
1.3.2 异相界面构筑 化合物间具有独特物理和化学性质的异相界面,能够对催化剂活性实现精细调控,因而越来越受到研究者们青睐。富含过渡金属氧化物与金属界面的Fe2O3/Pd、WO3/Pd以及MoO3/Pd 三种材料[88][图4(a)],Pd 在界面上的化学状态不同(+2~+3)[图4(b)],其中,Pd/Fe2O3界面上的Pd为+3价,当析氧电流密度为10 mA·cm−2时,过电位仅为383 mV[图4(c)]。通过液相还原法在NixFe3−xO4/Ni中构筑氧化物与金属界面[37],当催化剂中x 值为0.36时,析氧电流密度为10 mA·cm−2时仅需225 mV过电位。利用MOF−74 可控热解制备的富含NiCo/Fe2O3界面的催化剂[89][图4(d)、(e)],界面处NiCo 纳米颗粒能有效促进Fe 位点上*O 的形成[图4(f)],对提高析氧性能非常有利,当析氧电流密度为10 mA·cm−2时,过电位仅为238 mV,Tafel 斜率仅为29 mV·dec−1[图4(g)、(h)]。在Ni(OH)2/CeO2界面处[图4(i)],发现了电子由CeO2向Ni(OH)2转移的现象[90]。因此,当提高CeO2含量时,催化剂中活性高的Ni3+含量也会随着增加,当Ni(OH)2与CeO2的比例为4∶1 时,在1.0 mol·L−1KOH 中 过 电 位 仅 需220 mV 就 能 达 到10 mA·cm−2的析氧电流密度[图4(j)],Tafel 斜率为81.9 mV·dec−1[图4(k)][90]。对FeOOH/CeO2界面的研究也得到了相似的结论[91]。利用ZIF−67构筑出CeOx/CoS界面,发现CeOx的量决定着CoS 中Co2+和Co3+的比例,进而决定催化剂的析氧活性[92]。CeO2/CoS2界面上也存在类似的现象[93]。
1.3.3 缺陷位调控 缺陷位在晶体中广泛存在,氧空位与阳离子空位是常见的两种缺陷位。缺陷位邻近的原子具有悬挂键和空电子轨道,成为析氧反应中独特的活性位点。本部分将重点阐述利用氧空位和阳离子空位增强过渡金属氧化物析氧催化活性的策略。
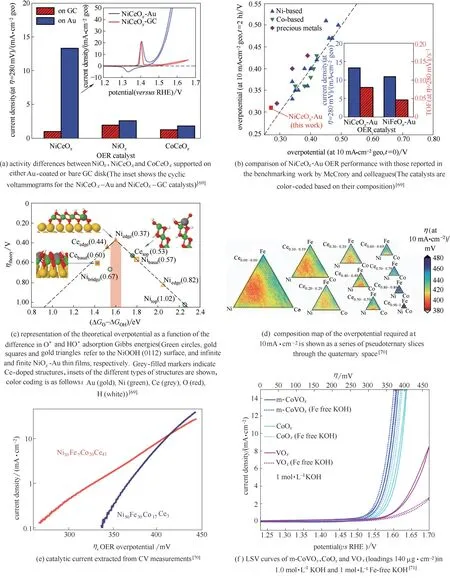
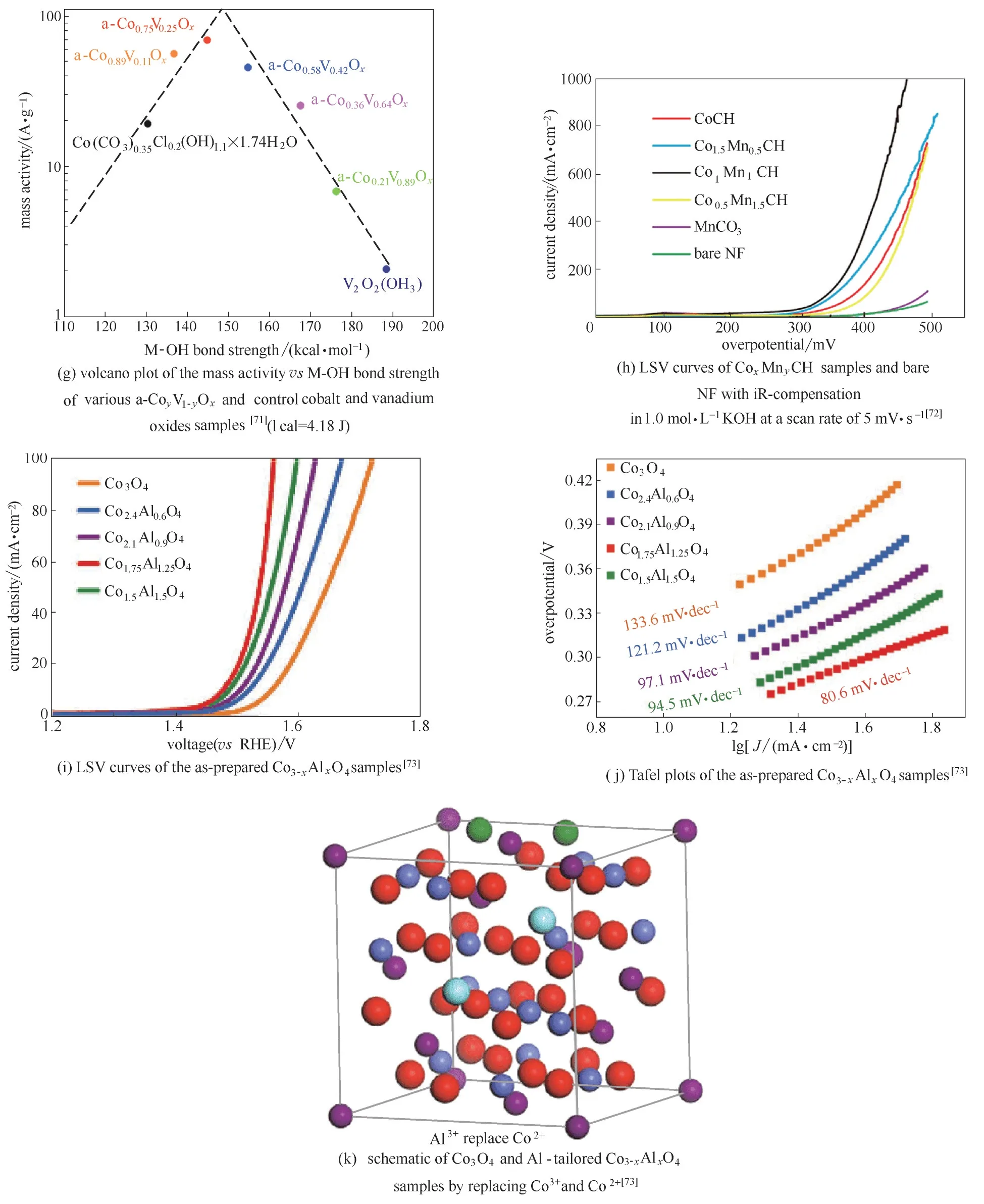
图3 杂原子掺杂增强过渡金属氧化物(TMOs)的析氧活性Fig.3 Heteroatom doping for enhancing OER activity of TMOs
(1)氧空位:过渡金属氧化物晶体结构中的氧,在特定条件下易脱离晶格,造成晶格中氧缺失,形成带正电的氧空位。氧空位在过渡金属氧化物中能降低带隙,提高HOMO 能级和费米能级,显著提升催化剂的析氧活性[94−95]。富含氧空位的催化剂可由多种方法制备。比如,通过氧等离子体刻蚀法制备的富含氧空位的Co3O4纳米片[96][图5(a)],晶体内氧空位的形成使部分Co3+被还原成Co2+[图5(b)],催化剂面积比活性因此提高了10 倍[图5(c)]。采用“吸附−煅烧−还原”策略,可制备富含氧空位的空心尖晶石型氧化物[45]。其中,珍珠链状NiCo 尖晶石型氧化物(R−NCO)中存在着大量氧空位,析氧反应的速率控制步骤也因此发生了改变[图5(d)~(f)],当析氧电流密度为10 mA·cm−2时,过电位仅为240 mV[图5(g)],Tafel 斜率仅为50 mV·dec−1[图5(h)][45]。利用多元醇配位还原法可制备富含氧空位的Co 纳米带[97]。利用液相还原法,可制备富含氧空位的FeCo双金属超薄纳米片FexCoy−ONSs(x 与y 代表Co 和Fe 的摩尔比)[98][图5(i)],纳米片厚度约1.2 nm,比表面积达到261.1 m2·g−1,氧空位有利于提高催化剂的电子传输与水分子吸附能力[图5(l)]。当x = y =1 时,FexCoy−ONSs 在析氧过电位为350 mV 时,质量比活性达到了54.9 A·g−1[图5(j)],Tafel 斜率低至36.8 mV·dec−1[图5(k)][98]。

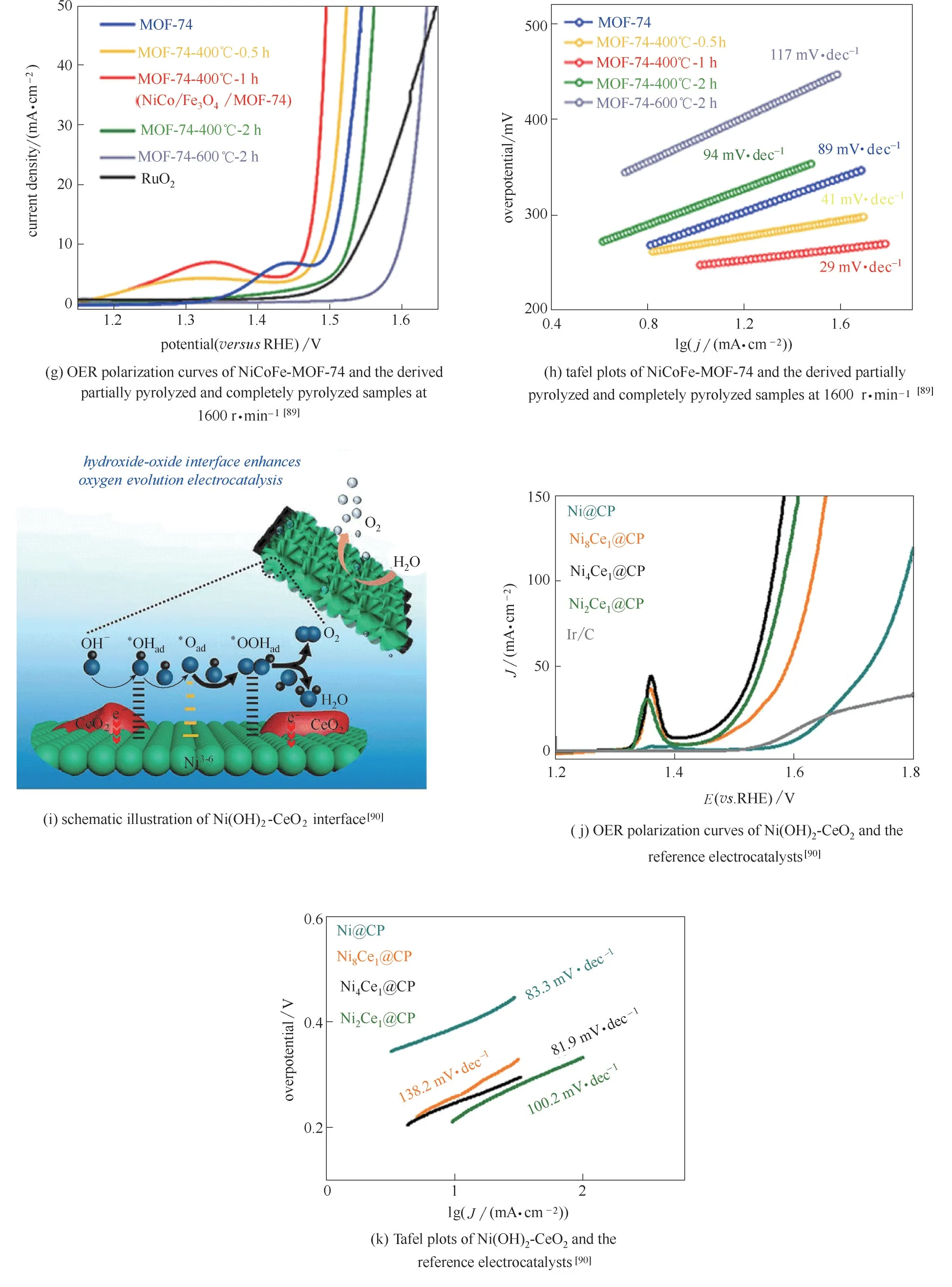
图4 构筑异相界面增强OER活性Fig.4 Constructing heteroatom phase for enhancing OER activity


图5 氧空位增强OER活性Fig.5 Oxygen vacancies for enhancing OER activity
(2)阳离子空位:作为一种很重要的缺陷位类型,阳离子空位也被利用来设计高活性的析氧催化剂。由于晶体整体呈中性,阳离子空位会导致邻近金属离子化学价上升,使催化剂析氧活性增加。Jaramillo 课题组[99]最早发现阳离子空位能促进析氧反应。当SrIrO3薄膜表面Sr 被刻蚀后得到IrOx/SrIrO3,在催化剂表面形成阳离子空位,使IrOx/SrIrO3在0.5 mol·L−1H2SO4中具有极高的析氧活性[图6(a)][99]。为探究阳离子空位在析氧反应中的作用,张强课题组[100]利用p区金属(同时具有金属性和非金属性)制备了系列富含阳离子空位的钙钛矿型氧化物[图6(b)]。以Sn4+为例,首先制备了NiFeSn 三元钙钛矿化合物,Sn4+在随后的析氧反应过程逐渐溶解,得到富含阳离子空位的NiFe 氧化物[图6(c)~(e)],在析氧电流密度为10 mA·cm−2时,过电位仅为350 mV[图6(c)][100]。王双印课题组[23]利用Ar 等离子刻蚀法制备了系列富含Sn 空位的钙钛矿型催化剂(表示为SnCoFe−Ar,图6(f)为刻蚀前后的TEM 图像)。样品SnCoFe−Ar 在析氧电流密度为10 mA·cm−2时,过电位仅为300 mV[图6(g)]。该课题还利用同样的方法制备了富含Fe 空位和Ni 空位的析氧催化剂[23][图6(h)]。

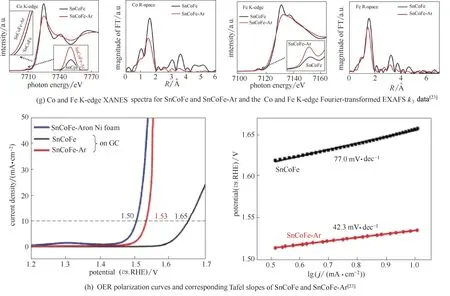
图6 阳离子空位增强OER活性Fig.6 Cation vacancy for enhancing OER activity
1.3.4 结构无定形化 无定形过渡金属氧化物内部金属原子和氧原子是以短程有序的形式排列,各类金属原子总体上分布均匀[101]。无定形化处理对提升过渡金属氧化物析氧活性也非常有效[102−105]。由于金属原子和氧原子具有很强的长程有序化排列的趋势,为无定形过渡金属氧化物的制备带来了较大困难[101]。
Shao−Horn 课题组[106]研究发现,钙钛矿型氧化物在催化析氧过程中,催化剂表面会发生重组,形成一层含有高价金属离子的无定形金属氧化物,其中的高价金属离子能激活催化剂的晶格氧过程,对增强析氧活性效果非常显著。Berlinguette 课题组[107]首次报道了通过光化学途径制备无定形金属氧化物的普遍化策略[图7(a)],得到的无定形金属氧化物几乎都拥有比晶态氧化物更高的反应活性[图7(b)、(c)]。由此法制备的Fe、Co、Ni 三元无定形金属氧化物,当Fe∶Co∶Ni 的摩尔比为2∶2∶1 时,析氧活性达到最高[108][图7(d)、(e)]。Zhang等[109]利用溶胶−凝胶法制备的CoFeW 三元无定形金属氧化物[图7(f)~(h)],在析氧电流密度为10 mA·cm−2时,过电位仅为191 mV,且长期稳定性超过500 h[图7(i)]。Lin 课题组[110]利用Cu+与S2O32−之间配位作用较强,Cu2O 中的Cu+能与Fe3+、Co2+、Ni2+等离子发生交换反应,以Cu2O 为模板制备了无定形的Cu−Ni−Fe 金属氧化物纳米笼[图7(j)],在析氧电流密度为10 mA·cm−2时,过电位仅为224 mV[图7(k)][110]。Shao课题组[111]则利用FeCl3的腐蚀作用,以LaNiO3为模板制备出无定形的钙钛矿金属氧化物,析氧电流密度为10 mA·cm−2时,过电位仅为189 mV。
1.3.5 形貌调控 上面四种析氧活性增强策略都是对电子结构或活性中间物种吸附自由能的调控,在此基础上进一步调控形貌,增加催化剂比表面积,最大限度地暴露催化剂活性位点,也是提高过渡金属氧化物析氧活性的必要手段。由于析氧反应涉及氧气泡析出,气泡在催化剂表面生成、长大以及脱离的过程中,会对催化剂产生拉应力。这种拉应力会导致催化剂从集流体上脱落,造成催化剂失活,在集流体表面构筑催化剂原位生长的一体化电极能有效减弱这种趋势,本节将主要论述一体化过渡金属氧化物电极中催化剂形貌的调控策略。


减小催化剂颗粒尺寸,可以增加催化剂比表面积、提高电化学活性。Nam 课题组[112]利用液相氧化法,通过精确控制反应条件,成功合成出直径分别为4 nm 和8 nm 两种尺寸的Mn3O4纳米颗粒,测试表明颗粒尺寸为4 nm的Mn3O4具有更高的比表面积和电化学活性,在0.5 mol·L−1PBS 中,析氧电流密度达到10 mA·cm−2时,仅需384 mV的过电位。

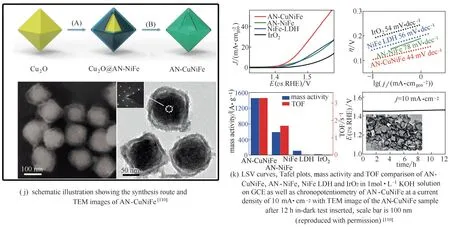
图7 无定形金属氧化物增强OER活性Fig.7 Amorphous metal oxide for enhancing OER activity
二维材料具有超大比表面积以及边缘位点暴露充分等优点,被视为一种理想的电催化剂材料[115]。然而,二维材料在使用过程中容易坍塌堆叠,掩盖活性位点,阻碍电解质传输,降低催化剂活性。Zheng课题组[116]通过阳离子交换法在Cu2O纳米线上生长出Ni−Co 氧化物纳米片[图8(c)],Cu2O 作为主体结构支撑具有较高析氧活性的Ni−Co氧化物纳米片,催化剂活性和稳定性同时得到提高。Wu等[117]利用多孔阳极氧化铝(AAO)为模板,制备了由厚度为2.4 nm 的Fe−NiOx纳米片组装而成的纳米管[图8(d)],当析氧电流密度为10 mA·cm−2时,过电位仅为310 mV,Tafel斜率49 mV·dec−1。
通过形貌调控优化催化剂的电子传输途径也是提升析氧活性的一种有效方法。Li课题组[50]总结了石墨烯负载的过渡金属氧化物用作析氧催化剂的研究,并提出石墨烯用作催化剂载体能显著提高其OER性能。Lyu等[33]利用具有金属导电性的MoO2材料,构筑出MoO2−CoO 杂化材料纳米笼[图8(e)],提高了析氧催化活性。此外,Yan 课题组[118]利用具有较强电子传导能力的Cu 构筑出Cu@CeO2@NiFeCr纳米线,优化催化剂的电子传导路径,降低电阻,提升析氧活性[图8(f)]。碳材料是一种更为经济、导电性更加优异的材料。然而,由于化学性质的差距,构筑过渡金属氧化物−碳材料复合催化剂难度较大。为此,Qiao 课题组[119]利用热解原位生长的Co−MOF 纳米线前体,制备得到多孔的Co3O4−C 纳米线复合催化剂,引入碳材料提高了催化剂的电子传导能力,降低了过电位[图8(g)]。
1.4 小结
本章节总结了近年来提高过渡金属氧化物析氧活性的主要策略(表1)。现阶段,过渡金属氧化物催化析氧反应的过电位依旧较高,高达数百毫伏,这主要是由于析氧反应包含复杂的4 电子转移过程和3 种活性中间物种的吸附。针对更高活性过渡金属氧化物析氧催化剂的开发,需要找到优化析氧反应各活性中间物种在催化剂表面吸附自由能的有效方法,同时进一步提高析氧催化剂的电子传导能力,降低催化剂与集流体间电子的传输能垒。
2 过渡金属氧化物析氧稳定性研究

图8 形貌调控增强OER活性Fig.8 Morphology control for enhancing OER activity
析氧催化剂长时间保持高活性对电解水制氢技术实现工业化应用非常关键。析氧催化剂涉及机械稳定性和化学稳定性。在析氧过程中,催化剂表面会产生大量气泡,气泡在成核、长大、滑移、聚并及脱离界面逸出过程中,会对催化剂表面产生较强的拉应力,造成催化剂从电极表面剥落,导致催化剂失活[54]。制备三维连通有序一体化电极能有效解决析氧催化剂机械稳定性问题,本部分将对过渡金属氧化物在化学稳定性方面的研究进展进行阐述。
2.1 失活机理研究
稳定性是过渡金属氧化物析氧催化剂研究中必须关注的问题。现代材料分析测试技术的飞速发展,为析氧催化剂失活机理研究提供了有力的工具和手段。目前,此领域的研究尚处于起步阶段,得到的初步结论为:无论贵金属Ir、Ru 还是非贵金属Ni、Fe 氧化物,析氧失活主要由活性金属组分溶解造成。
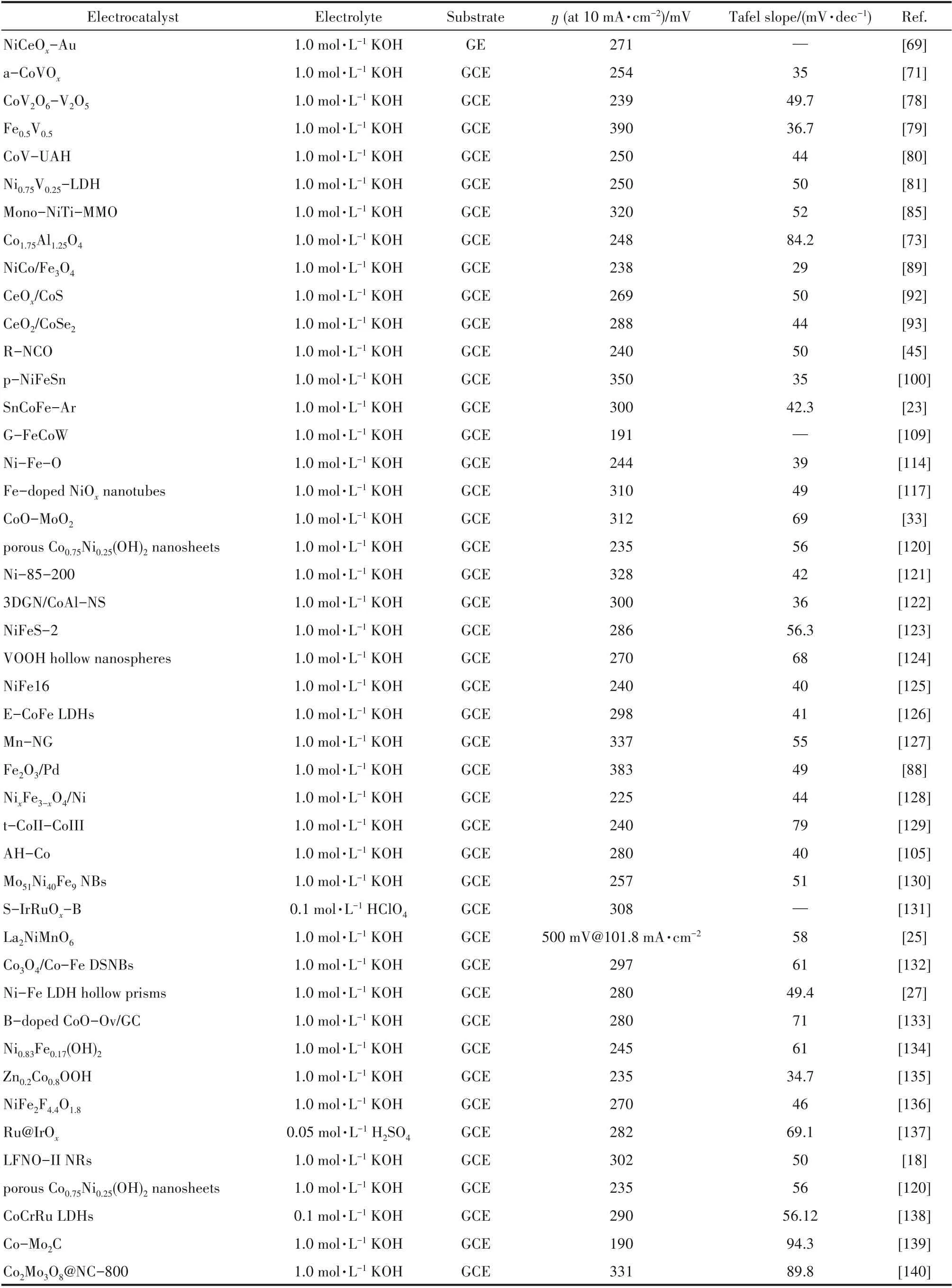
表1 过渡金属氧化物电催化析氧性能Table 1 Performance of transition metal oxides for oxygen evolution electrocatalysis

续表1
析氧催化剂稳定性研究始于对贵金属催化剂失活过程的探究,Cherevko 课题组和Markovic 课题组,在这方面做出了重要贡献。Cherevko 课题组[34]将流动电解池与电感耦合等离子体技术相结合,发现无论是在酸性还是在碱性电解液中,贵金属RuO2与IrO2都存在一定程度的溶解,且RuO2的溶解速率更大。因此,Cherevko 课题组得出,相较于RuO2,IrO2是一种更理想的析氧催化剂[34]。X 射线光电子能谱和同步辐射技术研究表明,金属Ir 向IrO2转变过程中形成可溶的Ir3+,是导致金属Ir稳定性差的主要原因,晶态IrO2在析氧过程中形成IrO42−则是造成晶态IrO2失活的主要原因[159][图9(a)]。
Cherevko 课题组[160]还深入对比研究了晶态IrO2、非晶态IrOx、钙钛矿型SrIrO3以及双钙钛矿型Ba2PrIrO6催化剂在酸性介质中稳定性和活性的关系。这几种催化剂活性的变化存在以下规律:Ba2PrIrO6>SrIrO3= IrOx>IrO2,而稳定性变化顺序正好相反[图9(c)]。XPS 结果表明,在Ba2PrIrO6、SrIrO3、IrOx三种催化剂表面形成了悬挂状态的IrO6八面体[图9(b)]。同位素示踪研究发现,在晶态IrO2表面产生的氧气,主要来自水分子,没有晶格氧过程发生,而在其余3 种催化剂表面检测到了16O18O和18O18O 信号,表明这3 种催化剂有晶格氧过程发生。结合催化剂活性、稳定性以及同位素示踪法研究,揭示出Ir 基催化剂表面悬挂状态的IrO6八面体在析氧过程中存在双重作用:(1)能促进晶格氧过程,提高IrOx的析氧活性;(2)会促进催化剂溶解,造成催化剂失活[图9(d)]。Cherevko课题组[77]将每溶解1 mg Ir 所对应的氧气析出量,定义为稳定性常数(用S 表示),来表征催化剂稳定性和预测催化剂的寿命。
在析氧过程中,过渡金属氧化物几乎都会被氧化成高价的氢氧化物。因此,研究过渡金属氢氧化物析氧稳定性有助于揭示过渡金属氧化物析氧失活机理。Berlinguette 等[161]最早发现,NiFe 氢氧化物在碱性电解液中催化析氧反应时,Fe 元素会逐渐溶解,造成催化剂失活。定量分析表明,即使析氧电流密度为10 mA·cm−2时,NiFe氢氧化物中的Fe元素也拥有显著的溶解速率。随后,Markovic 课题组[162]在Pt(111)晶面上沉积了一层MOxHy(M=Ni、Co、Fe)制备出三种模型催化剂,并深入研究了非贵金属氢氧化物在析氧过程中的活性变化机制[图9(e)]。三种催化剂析氧活性初始顺序为:NiOxHy<CoOxHy<FeOxHy,但FeOxHy的 溶 解 速 率(12.1 ng·cm−2·s−1)比CoOxHy与FeOxHy(分别为0.023 和0.004 ng·cm−2·s−1)高三个数量级[图9(f),(h)和(j)]。在1.7 V下连续电解1 h 后,FeOxHy的催化活性损失最大,NiOxHy活性几乎没有损失[图9(g)]。在MOxHy中掺杂Fe 可以进一步提升析氧活性,但Fe在析氧过程中的溶解又会降低MOxHy的稳定性[图9(i)],且活性提升越大,稳定性变得越差[图9(k)]。
Cherevko 课题组和Markovic 课题组的研究确认,无论是非贵金属Ni、Fe还是贵金属Ir、Ru氧化物的失活问题,都主要由析氧过程中活性金属组分溶解导致。
2.2 稳定性提升策略
现代材料分析技术的发展,使研究者们能够在析氧工况条件下深入研究催化剂表面的化学变化。近年来的初步研究表明,在工况条件下生成可溶的高价金属氧化物,是过渡金属氧化物析氧催化剂失活的主要原因。尽管有相当数量的过渡金属氧化物被报道在析氧条件下是稳定的,但还未建立起催化剂的“结构−稳定性”对应关系。另外,由于对活性组分溶解速率的报道不多,目前还难以严格比较各种析氧催化剂的稳定性。本章节将初步论述过渡金属氧化物析氧稳定性的提升策略。
2.2.1 平衡催化剂溶解与沉积速率 由于析氧催化剂失活主要是由活性金属组分溶解造成的,如能提高活性金属组分的反向沉积速率,则可以保持甚至提高催化剂的析氧活性。Markovic课题组[162]发现在电解液中添加0.1×10−6Fe 离子,Fe 离子会选择性沉积到NiOxHy表面,形成纳米岛形貌,有助于提升NiOxHy的析氧活性。对CoOxHy催化剂做同样处理,也观察到类似的现象。此外,当将Fe 掺杂的NiOxHy电极置于含有Fe 离子的电解液中电解时,Fe−NiOxHy不仅析氧活性高,稳定性也好[图10(a)]。通过同位素标记显示,57Fe−NiOxHy电极中57Fe 的溶解速率与溶液中56Fe 在电极上的沉积速率几乎相等,溶解−再沉积达到平衡[图10(b)~(d)]。定量计算表明,1 min 内约有50%57Fe 与溶液中的56Fe 发生了交换反应,1 h内约交换了70%。理论计算表明溶液中的Fe 离子容易沉积到NiOxHy表面,形成高活性的析氧反应中心。Fe在NiOxHy、CoOxHy、FeOxHy三种电极表面沉积的趋势为:NiOxHy>CoOxHy>FeOxHy。在实际操作中,可通过在电解液中添加Fe离子来同时提升过渡金属氧化物的析氧活性和稳定性。
2.2.2 构筑核−壳结构 通过核−壳结构的构筑,在析氧活性高的催化剂核外包覆稳定的保护壳层,能有效提升析氧催化剂的稳定性。Qiao 课题组[137]报道了一种核−壳结构的Ru@IrOx催化剂[图10(e)~(g)]。Ru 在催化剂中的化合价存在以下变化规律:RuO2>RuIrOx>Ru@IrOx>Ru foil,而Ir 的化合价变化规律为:IrO2>Ru@IrOx>RuIrOx>Ir。因此,Ru和Ir在Ru@IrOx核−壳结构中存在电荷的再分布,使Ru的化合价降低而Ir 的化合价升高,这有利于提升催化剂的析氧活性和稳定性。在析氧过电位为330 mV 时,Ru@IrOx的质量比活性达到644.8 A·g−1,分别是RuIrOx、RuO2NPs 和IrO2NPs 析氧质量比活性的3.7、5.9 和14.8 倍[图10(e)]。与RuIrOx相比,Ru 和Ir的化合价在Ru@IrOx中变化较小,因此,在0.05 mol·L−1H2SO4溶液中,Ru@IrOx中的Ru 和Ir 溶解速率远低于在RuIrOx中Ru 和Ir 的溶解速率[图10(f)],具有更高的稳定性。在NiFe 氧化物表面覆盖CeOx薄膜形成核−壳结构[图10(h)~(j)],由于CeOx对OH−、甲醇分子和乙醇分子具有选择性透过的能力,异丙醇等大分子无法透过,NiFe 氧化物的析氧稳定性也得到提升[163]。


图9 过渡金属氧化物在析氧过程中的溶解机理Fig.9 Dissolution mechanism of TMOs during OER process

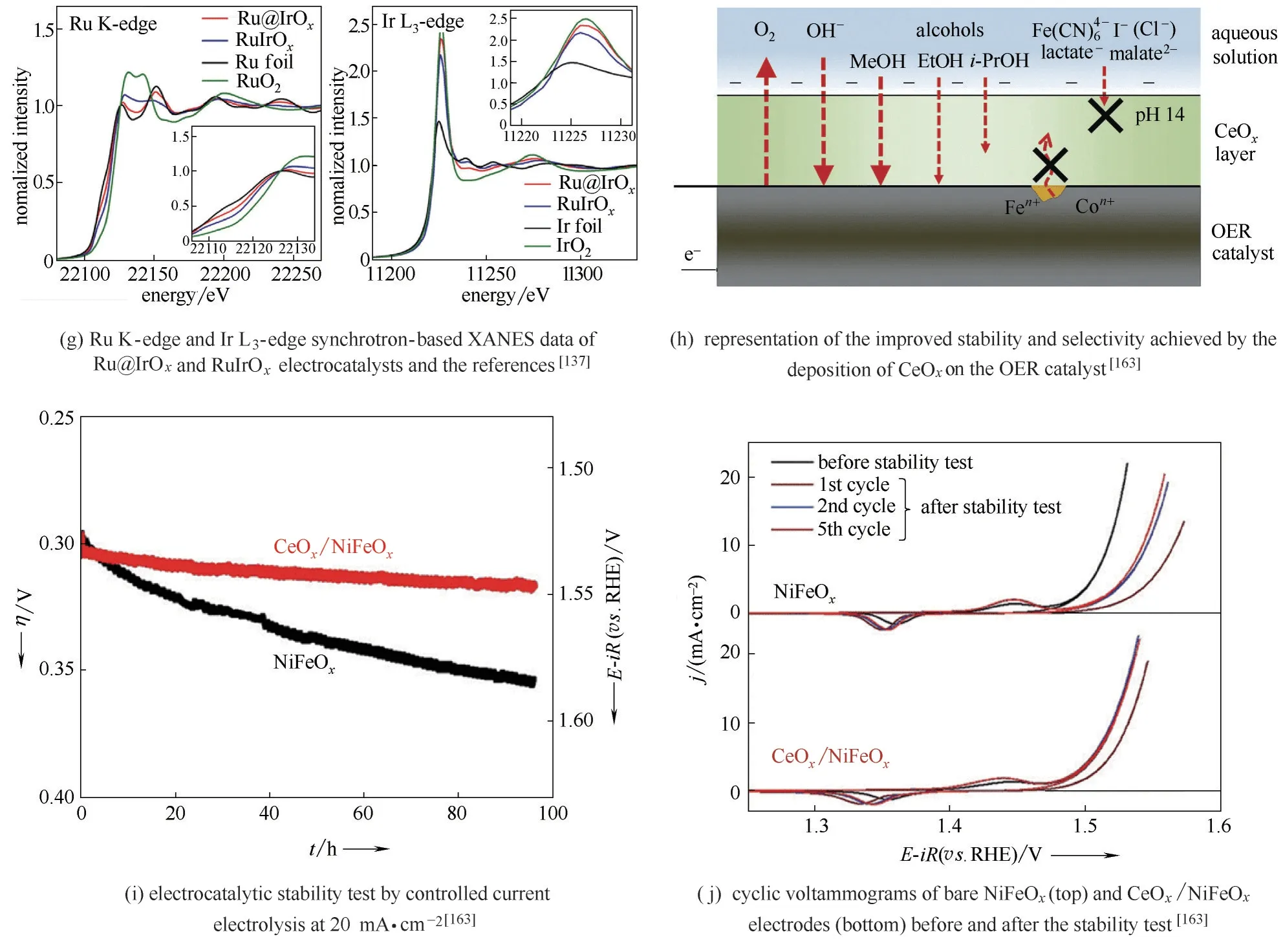
图10 增强过渡金属氧化物稳定性的策略Fig.10 Strategies for enhancing OER electrocatalysts stability
2.3 小结
本节阐述了近年来在过渡金属氧化物稳定性方面的研究进展。初步的研究表明,过渡金属氧化物活性金属组分在析氧过程中逐渐溶解是稳定性降低的主要原因,但具体的失活过程机理尚不明晰,仍需借助先进的原位探测手段进一步研究。在催化剂核外构筑稳定的保护壳层有助于提高催化剂的稳定性,但需以保障催化剂活性充分发挥为前提。此外,在电解液中添加Fe 离子,对提升特定催化剂的析氧活性和稳定性有利,具有“一石二鸟”的效果。然而,在实际电解装置中,Fe 离子的加入是否会对离子交换膜产生不利影响必须加以考虑。
3 结论与展望
本文总结分析了近年来在提升和增强过渡金属氧化物析氧活性和稳定性方面的研究进展。研究者们做了大量的工作,得到了许多有价值的结果。然而,过渡金属氧化物的析氧性能离工业化广泛应用仍存在一定距离,这主要由以下两个因素造成的:(1)从反应机理来看,析氧反应过程复杂,涉及4 电子转移和3 活性物种,反应过电位常常高达数百毫伏。(2)在电催化析氧反应过程中,催化剂表面金属元素易被氧化成高价金属离子而溶解到电解液中,导致催化剂稳定性变差。因此,未来在过渡金属氧化物用作析氧电催化剂研究中,应注重以下几个方面。
(1)过渡金属氧化物表面金属元素在析氧过程中与析氧反应前的化学状态有很大差异,如何精确调控表面金属元素的化学状态以获得更高活性是下一阶段研究的重点。
(2)缺陷位在过渡金属氧化物中广泛存在。然而,现阶段还缺乏针对过渡金属氧化物缺陷位的系统性研究,特别是缺乏缺陷位对析氧过程中催化剂表面金属元素化学状态及溶解速率影响的系统研究。
(3)过渡金属氧化物在酸性介质中的电催化析氧活性远低于在碱性介质中的活性。加强对酸性介质中析氧活性机理的研究,是开发酸性环境中高活性过渡金属氧化物析氧催化剂的关键。
(4)酸性介质腐蚀性强,限制了催化剂材料的选择。已开发的贵金属基催化剂(RuO2或IrO2)以及部 分 非 贵 金 属 氧 化 物(MnO2,CoFePbOx以 及NixMn1−xSb1.6−1.8Oy)在酸性介质中的催化活性依旧远低于工业化的要求。因此,进一步开发酸性介质中稳定的、活性高的过渡金属氧化物析氧催化剂是下一阶段的研究重点。
(5)现阶段,水热法是催化剂制备普遍采用的方法,由此法制备的一体化电极中催化剂与基体之间结合力依旧较弱,不能完全解决催化剂的机械稳定性问题。因此,需要研究新的催化剂制备策略,提高催化剂与集流体之间的结合力,以进一步提高过渡金属氧化物在析氧过程中的机械稳定性。
(6)当前,虽然对过渡金属氧化物在催化析氧反应过程中的失活机理有一定认识,但对确切的失活机理过程缺乏深入理解。借助先进的原位检测手段研究析氧过程中催化剂表面化学状态的变化,揭示过渡金属氧化物失活的具体过程,是开发兼具高活性和高稳定性的过渡金属氧化物析氧催化剂的研究重点。
