1 岁半起病的SGCE 肌阵挛-肌张力障碍1 例临床特征和基因分析
2020-09-25牟常华王纪文周昀箐王英燕贺影忠王翠锦姚如恩
牟常华 王纪文 周昀箐 王英燕 贺影忠 王翠锦 姚如恩
国家儿童医学中心 上海交通大学医学院附属上海儿童医学中心(上海 200127)
肌阵挛-肌张力障碍(myoclonus-dystonia,M-D)是一种运动障碍,其特征是快速、短暂的肌肉收缩和/或持续的扭转和重复的运动,约50%由位于7q21.3上编码ε-肌糖(epsilon-sarcoglycan)蛋白的SGCE基因变异所致。SGCE肌阵挛-肌张力障碍(SGCE-M-D)又称为肌张力障碍11型(dystonia 11,DYT11)或肌张力障碍-SGCE(DYT-SGCE),既往用于SGCE-M-D的其他术语包括遗传性肌阵挛-肌张力障碍综合征(inherited myoclonus-dystonia syndrome)和酒精反应性肌阵挛性肌张力障碍(alcohol-responsive myoclonic dystonia)等[1]。目前国外已有多个M-D家系或病例的研究报道,而国内则罕见报道[2]。现报告1 例1 岁半起病的SGCE-M-D患儿,对其临床表现及基因突变情况进行分析,并复习国内外相关文献,以期提高对SGCE-M-D临床诊治的认识水平。
1 临床资料
患儿,男,3 岁,因行走不稳和反复肢体抽动1 年半余于2019年10月17日在上海市儿童医学中心神经内科住院治疗。约1年半前患儿出现行走不稳,走路、跑步时易摔倒,下肢着地,意识清晰,可迅速站起,头部、躯体及皮肤等部位无伤痕。另外,患儿还出现反复肢体抽动,表现为下肢或上肢伴或不伴躯体抽动,双侧或右侧明显,部位不固定,瞬间恢复,有时间歇数秒至数分钟连续数下,抽动间歇期意识无改变,有时1天数次单下抽动。可在数天内反复出现,有时间歇数天至1 月左右无发作。肢体抽动和行走不稳不同步。肢体抽动均在清醒时发生,入睡后消失。患儿上述症状无晨轻暮重和日间波动现象,无明显发作后疲劳和思睡等表现。外院头颅磁共振成像(MRI)、动态脑电图(包括发作时脑电图)、血尿代谢筛查、血液检查等均未发现明显异常。曾用左乙拉西坦、德巴金、硝西泮及多美巴(具体剂量不详)等治疗,效果不佳。患儿病情逐渐缓慢加重,出现持物不稳易掉,动作缓慢费力,进食时右手扶碗,左手拇食指夹勺不易抬起,姿势僵硬,不易将食物送入口中等症状。父母否认患儿起病前感染、接种疫苗、外伤、食物和药物中毒等病史,发病以来患儿智力、语言、情绪、性格和睡眠、食欲、大小便等无异常。
患儿系G2P1,母孕期无异常,足月顺产,产时曾因哭声微弱吸氧数分钟,后无异常,出生体质量 3 100 g,生后生长发育同正常同龄儿。父亲24岁,母亲26岁,均体健,非近亲婚配。弟弟2岁,体健。否认遗传疾病家族史,家族中无类似疾病史。
入院体格检查:体温36.8 ℃,脉搏110次/min,呼吸22次/min,收缩压/舒张压(SBP/DBP)90/62 mmHg,身高100 cm,体质量19 kg;神志清,精神反应可,面容无异常,语言流利,对答切题,高级皮质功能正常,四肢无畸形,活动可,面色正常,营养良好;心肺腹无异常。神经系统查体:脑膜刺激症阴性,膝踺反射正常引出,巴氏征阴性。共济失调:右手指鼻试验阳性,左手指鼻试验阴性,闭目难立征阳性。跟膝胫试验阴性。四肢肌力、肌张力正常。
实验室检查:血常规、生化、电解质、甲状腺功能、血沉、EB病毒DNA、巨细胞病毒DNA均正常。胸部正位片:两肺未见明显活动性病变。心电图:窦性心律,不完全性右束支传导阻滞,头颅MRI未见异常。视频脑电图包括发作时未见癫痫样放电。血尿代谢筛查未见明显异常。
考虑患儿疾病表型可能存在基因突变,经家属知情同意及医院伦理委员会审批,签署基因检测知情同意书,送第三方检验机构(北京迈基诺基因检测公司)行全外显子检测,报告显示:SGCE基因c.1037+1G>A剪接突变。经Sanger验证,患儿父母该位点为野生型,先证者为新发变异。见图1。
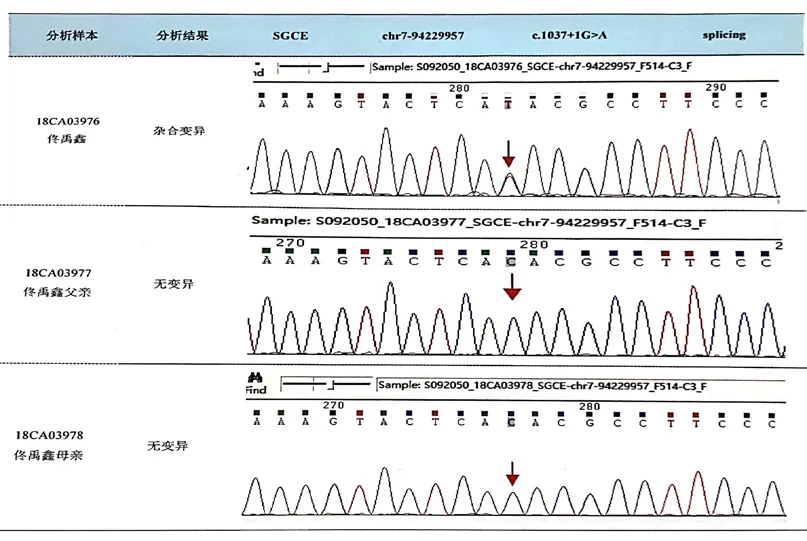
图1 患儿及其父母基因测序结果
患儿临床、基因检测结果均支持诊断为SGCE 肌阵挛-肌张力障碍,住院后逐渐逐个停服其他药物,服用唑尼沙胺早75 mg、晚50 mg [6.5 mg/(kg·d)],患儿仍有发作,表现为阵发性行走不稳,易跌倒,双上肢无力伴不自主抽动。神经外科会诊意见:暂时不宜行深部神经核刺激术。住院1 周后出院,出院后唑尼沙胺加量至早75 mg、晚75 mg [8 mg/(kg·d)]。出院后4个月随访,患儿症状较前好转,现可独走,偶有摔倒,右上肢持物稍欠稳,偶有肢体抽动。
2 讨论
肌阵挛是一种过度运动障碍,其特征是单个或多个肌肉突然、短暂和不自主地抽搐,可由肌肉收缩(正性肌阵挛)或目的等长收缩中肌肉活动的突然中断(负性肌阵挛)所引起。肌阵挛性抽搐有时难以与其他过度运动障碍进行区分,但电生理检查有助于两者的鉴别诊断,并可对肌阵挛亚型进行分类[3]。本例患儿18月龄左右出现发作性肢体抽动,需考虑癫痫的肌阵挛性发作和抽动障碍等非癫痫性发作。但患儿只有在清醒状态下发作,发作时和发作间期脑电图未见癫痫样放电,应用部分抗癫痫药物治疗无效等特点,可以排除癫痫。患儿起病年龄小,发作形式和部位固定,病情没有波动性等,不支持抽动障碍的诊断。肌张力障碍是一种间歇或持续性肌肉痉挛性收缩所导致的重复异常运动、姿势异常或两者均有的运动障碍疾病。主要特征为模式化的扭转可伴震颤,伴肌肉活动过度/溢出;其他特征包括任务特异性即某些主动运动诱发或加重、可存在缓解技巧(某些活动或某些部位触摸改善症状)、零点效应(某一种姿势或状态,肌张力暂时消失或明显缓解)和镜像现象(正常侧随意运动时可诱发患侧肌张力障碍)。上述特点均有助于区分肌张力障碍与其他运动障碍的表现[4-5]。本例患儿行走不稳,走路姿势异常,经常出现摔倒,主动摄食时手部动作异常,安静状态时肢体无异常,无眼球震颤、意向性震颤和语言障碍等症状和体征,智力发育正常,头颅MRI未见异常,不支持大脑、脑干、小脑、脊髓等神经系统的器质性病变,而符合肌张力障碍的特点。
最近国际帕金森和运动障碍疾病协会[3]提出了肌阵挛综合征和伴肌阵挛抽搐的相关疾病(过度惊吓反射和肌阵挛性癫痫性脑病)的命名,发现前者几乎均有其他运动障碍,其中包括共济失调、肌张力障碍和两者均有,但后者则无其他运动障碍。2013年肌张力障碍国际专家共识委员会更新了肌张力障碍的分类标准[4-5],将肌张力障碍根据是否合并其他运动障碍和其他神经系统表现(如认知功能减退、痉挛状态、耳聋等)或全身表现可分为单纯性、复合性和复杂性肌张力障碍,其中肌阵挛-肌张力障碍是一种复合性肌张力障碍,特点是肌阵挛与肌张力障碍的随机任意组合。近来有研究者提出,M-D 的诊断需要符合4 个主要标准和没有排除标准中的情况,或3个主要标准、2个次要标准和没有排除标准中情况[6]。其中主要标准包括:①单纯的或比肌张力障碍占主导地位的肌阵挛;②上身运动表现突出;③无躯干肌张力障碍;④阳性家族史;⑤18岁前起病;次要标准包括:①强迫症、焦虑相关性疾病或酒精依赖;②儿童或青少年期自发缓解的肢体肌张力障碍;③酒精反应性。排除标准:①除肌阵挛和/或肌张力障碍以外的其他神经系统表现(除癫痫发作,某些SGCE-M-D 患者可能存在癫痫发作);②脑MRI检查异常;③不支持肌阵挛诊断的神经生理学发现。许多学者认为“M-D”是用于具有SGCE样表型个体的术语,其区别于“肌阵挛性肌张力障碍(myoclonic dystonia)”,后者主要表现为肌张力障碍,在肌张力障碍身体区域同时伴有较长持续时间的抽搐[1,6]。目前发现,肌张力障碍合并肌阵挛的相关基因主要包括SGCE、CACNAlB、KCTDl7基因[7],此外三磷酸鸟苷环化水解酶1(GCH1)、酪氨酸羟化酶(TH)基因变异也可表现为肌张力障碍合并肌阵挛。此3种肌张力障碍合并肌阵挛症状均累及头颈、躯干和四肢,部分伴有焦虑、强迫、抑郁等精神症状,临床表现相似不易区分[7]。本例患儿主要表现为肌阵挛和肌张力障碍,SGCE发现致病性变异,基本符合以上的主要标准和没有排除标准中的情况,应该诊断为SGCE-M-D。
SGCE-M-D 发病通常在10 岁以内,且几乎总是在20岁之前,但生后6个月至80岁均可发病;病程进展缓慢,主要表现为肌阵挛、肌张力障碍和精神症状。典型的肌阵挛性抽搐最常影响颈部、躯干和上肢,下肢受累较少。约50%的患者有其他局灶性或节段性肌张力障碍,表现为颈部肌张力障碍如痉挛性斜颈和/或书写痉挛。肌张力障碍在病程中不倾向于恶化或泛化。受累身体部位的主动运动常可促发或加重不随意运动,其他诱发或增强因素包括压力、突然噪音、咖啡因和触觉刺激等。非运动性症状主要表现为强迫、焦虑、抑郁症状、惊恐发作、酒精滥用等。部分患者可有姿势性或其他形式的颤抖。罕见痴呆和共济失调等其他神经系统体征和症状[1-2]。SGCE-M-D 在婴幼儿中的临床表现罕见描述,有报道婴儿期起病的M-D可见下肢肌张力障碍,儿童下肢和躯干的肌阵挛偶可导致跌倒[8-9]。本例患儿起病年龄小,下肢受累运动障碍较明显,经常摔倒,没有痉挛性斜颈和明显的精神症状等特点,是否是婴幼儿发病SGCE-M-D 的临床特点,尚需进一步随访和大样本病例的研究。国内报道一个M-D家系中,1例先证者发病年龄为6岁,4年后才得以确诊[2]。因此对于此类患者,需详细询问病史和家族史、观察有无肌阵挛和/或肌张力障碍表现,尽早完善相关基因检测对诊断和治疗具有重要的指导意义。
SGCE-M-D 为常染色体显性遗传,其外显率取决于SGCE等位基因改变的亲本来源,具有母本印迹(maternally imprint)的特点,即父系衍生表达的SGCE等位基因的致病性变异通常会导致疾病,而母源性沉默的一般不会致病。目前已报道100多种SGCE致病变异,包括错义变异、移码变异、剪切变异、无义变异、大片段甚至整个外显子缺失等,大多数见于白种人。迄今为止尚未发现SGCE基因型和临床表型的对应性,相同的SGCE基因变异类型可具有不同的临床表型[1]。既往我国报道的SGCE基因变异类型和位点见图2所示,包括大陆地区的c.662+linsG位点的插入变异、c.237C>T位点的点突变、c.835_839delACAAA的缺失变异和台湾的c.842 delA 位点的缺失变异、c.524_531delTGGCCAGT位点的缺失变异、c.exon2-11del外显子的大片缺失[2]。本例患儿为东北地区的18月龄起病的SGCE-M-D,新生变异的SGCE基因位点为c.1037+1G>A剪接变异,导致氨基酸剪接改变,致使ε-肌糖蛋白的翻译受阻,从而影响该蛋白执行正常功能。本例患儿的变异位点尚未见报道。
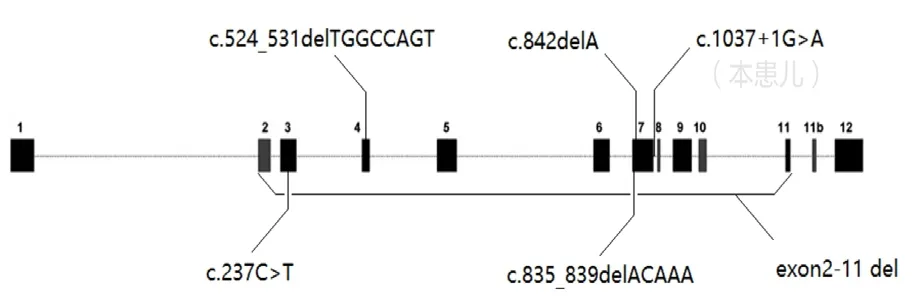
图2 本例患儿和既往我国报道的SGCE 基因突变位点
小脑-丘脑通路功能异常在肌阵挛-肌张力障碍的发病机制中至关重要。饮酒可以改善小脑相关的运动功能障碍,暂时增强GABA 能传导,提示GABA 能缺陷会引起浦肯野细胞功能异常[10]。除了小脑网络功能失调外,基底神经节和皮质病变也牵涉其中。在进行深部脑刺激手术时进行的微记录研究表明,患有肌阵挛性肌张力障碍患者的内部苍白球具有异常的神经元活动,其爆发更快和更短,其局部电场电位与受影响的肌肉活动之间的连贯性增强[11-12]。此外,血清素和多巴胺稳态的破坏也可能在发病机理中起作用。肌阵挛性肌张力障碍患者脑脊液中的血清素代谢物含量低,SGCE基因敲除小鼠中纹状体水平的内源性多巴胺增加,多巴胺D 2 受体表达降低或肌阵挛性肌张力障碍患者多巴胺D2受体利用率降低[13-16]。
目前有1级证据支持唑尼沙胺可改善SGCE-M-D的肌阵挛和肌张力障碍症状[17]。苯二氮卓类药物(特别是氯硝西泮)和用于治疗肌阵挛的抗癫痫药(尤其是丙戊酸和左乙拉西坦)也能改善肌阵挛,而其他抗癫痫药物如托吡酯等的效果不一致。抗胆碱药物可能会改善肌张力障碍。肉毒毒素注射可特别有助于缓解颈部肌张力障碍症状[1,18]。也有报道L-5-羟色氨酸、L-多巴和羟丁酸钠盐有效者。深部脑刺激可改善肌阵挛和肌张力障碍,大多数以内侧苍白球为靶点,但也有靶向丘脑腹内侧核成功者。大多数成年患者由于酒精的摄入,肌阵挛可急剧减少。虽然酒精摄入可短期改善症状,但由于成瘾的风险,不建议长期使用。SGCEM-D 可自发缓解,也可能逐渐进展,导致相当多的功能残疾,或影响患者工作和生活,但SGCE-M-D 患者的寿命通常不受影响[1]。本例患儿曾应用左乙拉西坦、丙戊酸、硝西泮及多美巴等药物,均效果不佳,应用唑尼沙胺后症状缓解明显,未行深部脑刺激。
