新发8p 重复伴缺失综合征患儿细胞分子遗传学研究
2020-09-25刘芙蓉郝胜菊周秉博
刘芙蓉 郝胜菊 张 钏 周秉博 王 兴 郑 雷
甘肃省妇幼保健院医学遗传学中心 甘肃省出生缺陷防控重点实验室(甘肃兰州 730050)
8号染色体短臂倒位重复伴末端缺失,即inv dup del(8p),是一种复杂的染色体质量组,目前报道新生儿中发病率为1/(10000~30000)[1]。2010年国内报道首例明确诊断病例[2]。此类患者共同的临床表征为智力障碍、脑部结构异常、特殊面容及先天性心脏病[3-6]。本文通过对1 例临床表型相似患儿的细胞分子遗传学分析,结合多种检测技术相互验证,明确病因,为临床诊断、预后评估及针对性遗传咨询提供有效依据。
1 临床资料
患儿,女,足月顺产,G2P1,出生体质量3 110g,无抢救史及异常出生史。6 月龄时于甘肃省妇幼保健院首次就诊。患儿就诊时体质量5.7 kg,身长64 cm,头围42.5 cm;面容异常,圆脸、高前额、眼球略凸出、睡觉不能闭眼、颞部收缩、鼻宽、鼻孔朝天、腭弓较高、人中长、下颌较小;对外界刺激反应缓慢,表情呆滞,少哭闹;俯卧位抬头<90°,四肢肌张力低下,特发性肌张力异常。心脏彩超提示:主动脉弓中断,室间隔缺损,房间隔缺损,动脉导管未闭。喉镜检查示:喉软化症。头颅CT平扫未见明显异常。
根据患儿临床表现,经医学伦理审查,患儿双亲知情同意后,采集患儿及双亲外周血,行细胞培养、染色体制备、G显带及染色体核型分析。结果显示,当分辨率在400 条带水平时,所有分裂相8 号染色体短臂8p21处增加一段不明来源的染色体物质,8号染色体短臂末端8p23处缺失条带,分析确定为8p臂内倒位所致的重复和缺失,但具体断裂点于当前分辨率下不易确定,见图1。患儿染色体核型为:46,XX,der(8)inv dup(8)(p21),del(8)(p23)患儿父母外周血染色体G显带核型分析均无异常。
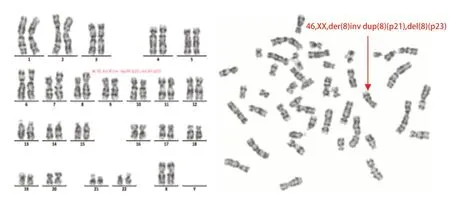
图1 染色体G 显带核型分析结果
使用QIAGEN 公司的基因组DNA 提取试剂盒,按照试剂盒说明书提取基因组DNA。Nanodrop2000核酸定量仪测定DNA 纯度和浓度。DNA 浓度范围为50~250 ng/μL。-20℃保存。将已构建的DNA文库在Illumina NextSeq500测序平台上机测序,最小精度为100 kb,测序深度为1 X。将高通量测序结果经信息分析,每个测序Reads匹配到其所在的染色体上作相应标准化Z值分析,通过Z值进行染色体异常判定。通过检索DECIPHER,OMIM,ClinGen,ClinVar,DGV等数据库确定CNV 的临床意义。选取断裂位点交界处基因,设计引物,由上海生工生物有限公司合成,检测位点序列及产物片段见表1。高通量测序全基因组拷贝数变异分析检出患儿8号染色体短臂存在100Kb以上已知的、明确致病基因组的拷贝数变异(CNV),其中在8p23.3-p23.1(160 001~7 120 000)区域,缺失6.96 Mb片段;在8p23.1-p21.1(12 560 001-27 940 000)区域,重复15.38 Mb片段,见图2。查阅文献,8p23.2-pter缺失患者主要的临床表征为智力障碍,发育迟缓,心脏异常等[6-7];8p22-8p23.1重复患者主要的临床症状为颅面畸形,特殊面容,骨骼、神经系统、心脏及泌尿生殖系统异常等[8-10]。
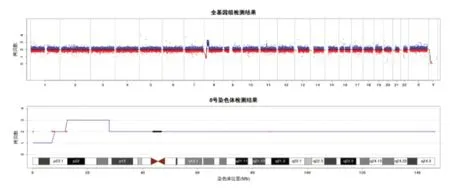
图2 高通量测序全基因组拷贝数变异分析结果
进一步以荧光定量PCR 检测技术验证CNV 结果。选取拷贝数变化区域的交界处,设计引物扩增验证。用GAPDH基因位点做内参,检测8p23.3-p23.1区域内基因组位置为6.90 Mb 的DEFA 5 和7.2 Mb的ZNF 705 G 基因位点拷贝数。PCR 反应总体系为20 μL,其中加入DNA模板40 ng,上下游引物各40 pmol,SYBR Green PCR Master2×预混液 10 μL。反应条件:37℃预变性5分钟,95℃预变性5分钟,95℃变性15秒,60℃延伸1分钟,共40个循环。利用ABI公司Quant Studio DX荧光定量PCR仪进行检测。数据分析以正常对照样本的待检基因和内参基因为基准进行校正,得出检测样本中待测基因和内参基因拷贝数比值的相对变化2-ΔΔCt[11]。靶序列体外PCR扩增时,荧光信号到达设定域值所需的循环数为Ct值;同一样本(先证者/正常对照)中扩增待检基因与内参基因的Ct差值为ΔCt;而待检样本与正常对照样本间ΔCt的差值为ΔΔCt。因此,正常对照样本的基因组拷贝数为2n,若检测样本基因拷贝数为1n则表示缺失1个拷贝,2n为拷贝数正常,3n为增加1个拷贝,相应的预期的2-ΔΔCt分别为0.5、1.0和1.5。结果显示,患儿与正常对照样本的比值分别为0.52和1.03,说明患儿上述位点的基因组拷贝数分别为缺失1个拷贝和无拷贝数变化。检测8p23.1-p22区域内基因组位置为12.3 Mb的FAM86B2和12.6 Mb的LONRF1的基因位点拷贝数,患儿与正常对照样本的比值分别为1.04和1.59,患儿上述相应位点的基因组拷贝数分别为无拷贝数变化和增加1个拷贝。检测8p21.1区域内基因组位置为27.6 Mb的ESCO2和28.2 Mb的PNOC基因位点拷贝数,患儿与正常对照样本的比值分别为1.51 和0.99,患儿上述相应位点的基因组拷贝数分别为增加1个拷贝和无拷贝数变化。荧光定量PCR与CNV检测结果一致,据此可确定染色体核型分析结果为,46,XX,der(8)inv dup(8)(p21.1-p23.1),del(8)(p23.1-p23.3)。

表1 荧光定量PCR检测位点各引物序列及产物片段长度
2 讨论
8 号染色体短臂的重复缺失绝大多数是倒置所致,其主要临床特征为:颅面畸形、特殊面容,如圆脸、前额突出、鼻梁塌、眼角下斜、高腭弓、小下颌、耳位及耳旋转度异常等,大脑结构异常,如胝体缺如、脑发育不全等,生长发育迟缓、智力发育落后,肌张力异常。另外,常可并发心脏异常,如卵圆孔未闭、右位心、动脉导管未闭、房室间隔缺损及法洛四联症等,骨骼异常及泌尿生殖系统异常等[12-15]。本例患儿的圆脸、高前额、鼻宽、小下颌等均与报道表型相符;与同龄正常儿童生长发育水平相比,患儿身长、体质量落后,且智力发育迟缓;特发性肌张力异常;并发房间隔缺损;虽头颅CT结果示大脑结构正常,但并未排除功能是否如常,仍有待进一步随访观察。综合患儿的上述各表型均符合inv dup del(8p)综合征的典型临床症状。
随着分子遗传学诊断技术的不断进步,对inv dup del(8p)综合征的研究逐渐深入,发现不同患者其8p末端缺失的片段完全一致,而倒位重复的片段则因双着丝粒中间体的断裂位点不同,致使近端断裂点各有不同[2]。有报道单纯8 p 末端缺失患者有圆脸、面颊饱满、突前额、耳位/耳廓异常、小下颌及先天性心脏病等临床表型特征,与inv dup del(8p)综合征临床表型相似[7]。因此8p23.1-8pter上相关基因的缺失导致出现inv dup(8p)综合征的特征性面容。本例患儿8p23.1至8p末端缺失,其特殊面容与报道相符,且心脏发育异常,提示单纯8p缺失其表型与8p重复伴缺失的表型类似。
研究显示,8p三体常伴胼胝体发育不良,且8p22-8p23.1片段重复导致胼胝体缺如、脑室扩张、脑发育不全等表型[16]。也有报道8p12-8p23.3倒位重复患儿,伴有显著的运动发育迟缓、肌张力减退、自闭症、面部畸形、室间隔缺损及胼胝体发育不良[17]。本例患儿重复片段包含至上述报道的区域内,其面容异常,圆脸,发育迟缓,特发性肌张力异常,先天性心脏病等特征均与报道相符,判定患儿符合inv dup del(8p)综合症的临床特征。利用染色体核型分析技术明确患儿8p臂内倒位,结合CNV技术对8p倒位重复及缺失片段进行精确定位,采用qPCR 技术选取断裂区域交界处基因位点进行验证,综合多种检测技术及患儿临床表现明确诊断为inv dup(8p)综合征。
目前inv dup(8p)综合征尚无有效治疗方法,产前诊断可减少此类患儿的出生。虽然本例患儿的父母染色体核型分析结果均无异常,但再次妊娠时仍有再发风险,建议通过产前诊断监测8p的异常重组,利用羊水细胞染色体核型分析、荧光定量PCR分析及CNV等多种细胞及分子遗传学检测手段,结合B超监测胎儿发育动态,对比临床表型明确胎儿状态。
今后对inv dup(8p)综合征的研究重点应为8p缺失、重复区域内相关基因的剂量效应与表型的相互关联及对相应基因功能的验证。
