先天性糖基化障碍Ie 型合并先天性肌营养不良表型患儿临床和基因分析
2020-09-25何小恋
祁 婧 逯 军 何 波 何小恋
1.中南大学湘雅医学院附属海口医院(海南海口 570208);2.海南省妇女儿童医学中心(海南海口 570206)
先天性糖基化障碍(congenital disorder of glycosylation,CDG)是一组由糖蛋白合成缺陷而导致的罕见先天代谢性疾病,大多为常染色体隐性遗传,少部分为常染色体显性遗传和X 连锁形式[1]。自1980 年首次临床描述以来,目前已发现130 种不同类型的CDG[1-3]。CDG 是具有临床异质性的多器官系统受累性疾病,尽管此类疾病的症状、体征和严重程度在不同的表型有所差异,但几乎任何器官系统都可能受到影响[4],最常见的表现是发育迟缓、肌张力低下、神经系统异常、肝病和凝血功能障碍。CDGIe 型为CDG 中的罕见类型,是DMP 1基因缺陷引起多萜醇磷酸甘露糖基转移酶1(dolichol-phosphate mannosyltransferase1,DPM1)活性下降,使多萜醇-磷酸-甘露糖(Dolichol-P-Mannose,DPM)复合物合成过程受损,主要累及神经及肌肉等器官系统。本文回顾分析1 例CDG-Ie 型合并先天性肌营养不良表型患儿的临床资料及基因检测结果。
1 临床资料
患儿,男,G3P3,足月顺产,Apgar评分10分,出生体质量3.65 kg,无窒息史及产伤史。患儿父母均健康,非近亲婚配,母孕期无异常;有两胞姐,大姐生后14天因小头、进食及吞咽困难死亡;二姐3岁,体健。患儿于1月龄体检发现头围32 cm。行头颅磁共振成像(MRI)提示头围偏小,脑外间隙弥漫性增宽,脑内髓鞘化明显偏弱(图1)。家属拒绝进一步检查。患儿于生后1 个月24 天因咳嗽3 天、发热半天就诊,无抽搐,无腹泻。查体:体质量3.7 kg,头围33 cm(<3SD),身长60 cm;神清,精神反应稍差,前囟平软,面容异常,双眼上斜、扁鼻梁、小下颌,呼吸急促,三凹征弱阳性,双肺呼吸音粗糙,可闻及固定湿啰音;心率135次/min,心律不齐,心音有力,未闻及心脏杂音;腹部无异常;四肢肌力、肌张力偏低,哭闹及活动时肌张力增高,双足呈挛缩状态,余神经系统未见异常。实验室检查:肌酸激酶2 238 U/L,乳酸脱氢酶429 U/L,肌酸酶同功酶75.0 U/L,纤维蛋白原1.33 g/L,部分凝血酶原时间45.60s,凝血酶时间25.20s,抗凝血酶31.5%;血常规、肝肾功能、大小便常规、脑脊液常规、生化、呼吸道病毒抗体四项等均未见异常;血串联质谱、尿有机酸检测未见异常。胸部CT提示双肺多发渗出,双侧胸膜增厚。

图1 患儿脑部MRI 表现
入院后予抗感染治疗。分别于入院第3、6、8天出现抽搐3次,均为无热抽搐,表现为强直阵挛样发作,予苯巴比妥钠后未再抽搐。患儿4月龄时再次出现无热抽搐,性质同前,约5、6次/天,追听、追视差,肌力及肌张力低下。家属拒绝实验室检查及住院治疗,急诊予以苯巴比妥钠止惊后。24小时动态脑电图示频繁出现周期性暴发抑制,高波幅段出现1.5~4 Hz复合慢活动,间杂尖慢波、棘波、棘慢波阵发性发放,持续3~4s,低波幅为广泛平坦电位,持续约7s左右,清醒期及睡眠期均有多量放电(图2)。考虑婴儿痉挛症予以泼尼松2 mg/kg口服,抽搐发作无缓解,更换为氨己烯酸片20 mg/(kg.d),每日2次口服,发作次数减少。
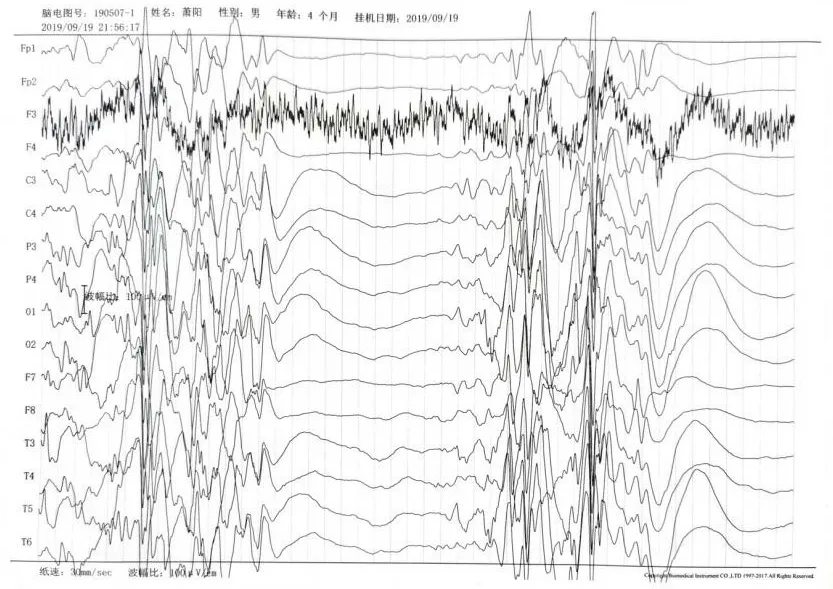
图2 患儿脑电图表现
随访至11 月龄时,患儿尚不能独坐、翻身,不能听懂自己的名字,语言、智力运动发育落后于同龄儿。经医学伦理审核及父母知情同意后完善基因检测。抽取患儿及父母外周血,送北京康旭医学检验所进行全外显子基因测序。结果显示,患儿DPM1基因存在复合杂合变异,分别为c.669-3C>G和c.677G>T点突变,患儿父母均为正常临床表型携带者,其中c.669-3C>G点突变来源于母亲,为编码区第 669 号核苷酸前内含子中倒数第3 位核苷酸由 C 变为 G 的杂合核苷酸变异,是剪切变异;c.677 G>T 突变来自父亲,为编码区第 677 号核苷酸由 G 变为 T 的杂合核苷酸变异,该变异导致第 226 号氨基酸由精氨酸变为亮氨酸(p.Arg226Leu),为错义变异。见图3。上述变异均可能导致蛋白质功能受到影响,均不属于多态性变化,在人群中发生的频率极低(参考数据库:1000Genomes、dbSNP)。
2 讨论
文献报道,欧美人群CDG患病率为1/10 000,其中最常见的类型为CDG-Ia型,CDG-Ia的患病率在荷兰人群中为1/20 000,在爱沙尼亚则为1/77 000[1],我国目前无发病率统计。此外,根据欧洲1 项调查,1 350例确诊CDG中,CDG-Ia占61.7%,其次为CDGIc 约为7.4%,CDG-Iq 3.1%,CDG-Ik 3.0%,CDGIb2.6%,未发现有CDG-Ie[5]。回顾国内外文献,迄今国内尚无CDG-Ie的病例报道,国外文献有8篇共9例CDG-Ie患者被报道。
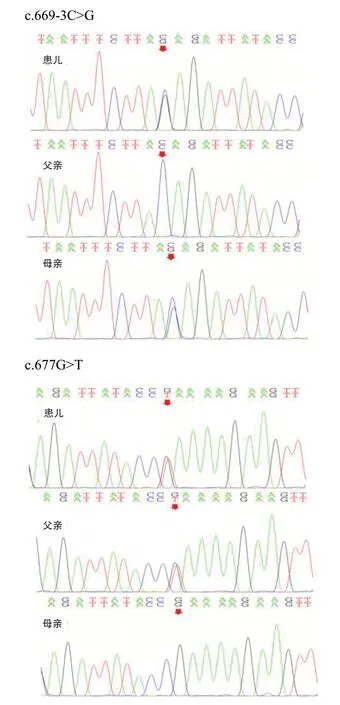
图3 患儿及父母DPM1 基因测序峰图
糖基化是在不同细胞途径中向蛋白质和脂质中添加糖残基的过程。CDG 可发生于糖基化的各个步骤,根据糖蛋白中糖基或糖链与蛋白质的连接方式,CDG包括(I型)N-连接糖基化、(II型)O-连接糖基化、(III型)N-和O-联合多重糖基化以及(IV型)脂质和糖基磷脂酰肌醇(GPI)锚定连接缺陷四种类型缺陷[1]。N-连接糖基化是一种翻译后蛋白质修饰,发生在内质网和高尔基体中,涉及一系列步骤,组装碳水化合物树,共价连接到天冬酰胺的酰胺基团及剪接。O-甘露糖基化是O-连接糖基化的一种亚型,涉及甘露糖聚糖与新生蛋白质的连接。CDG-Ie 型属于CDG 中较为罕见的一种,可同时合并N-连接糖基化和O-连接糖基化障碍,其临床表现多样,多以神经系统症状为主,合并α-抗肌萎缩相关糖蛋白(αdystroglycan,α-DG)的O-甘露糖基化障碍可表现为先天性肌营养不良表型[6]。CDG-Ie 型因DMP 1基因缺陷致DPM 转移酶活性下降而致病。DPM复合物是由3个蛋白质亚基(DPM1、DPM2和DPM3)组成,其中DPM1是细胞质催化亚基,位于20q13.13上,包含10个外显子,通过DPM3锚定于内质网膜,DPM2作用为稳定复合物[7]。DPM转移酶催化多萜醇-磷酸和GDP-甘露糖合成DPM,后者在各种糖基化过程中(包括N-糖基化和O-甘露糖基化,C-甘露糖基化和GPI锚定连接)作为供体底物,其缺陷将导致糖基化缺陷[8]。
α-DG是高度糖基化的蛋白质,在肌营养不良蛋白复合物中起着不可或缺的作用,其糖基化主要为人体内少见的O-连接糖基化,到目前为止哺乳动物的O-甘露糖聚糖仅发现存在于周围神经、骨骼肌和脑组织中,是高度糖基化的基底膜受体,该蛋白糖基化是受体功能和配体结合所必需的,通过它的多聚糖结构“基质聚糖”,与细胞外基质中含有层黏连蛋白球型(laminin globular,LG)结构域的配体,如层黏连蛋白、串珠素、集聚蛋白、轴突蛋白和皮卡丘素等结合,在维持肌细胞完整性和脑发育方面发挥重要作用[6,9-10]。由于编码糖基转移酶的基因变异,导致α-DG 的O-连接糖基化缺陷导致的一组疾病称为α-抗肌萎缩相关糖蛋白病(α-dystroglycanopathy,α-DGP)。除了CDG相关基因(DPM1、DMP2、DPM3、DOLK)外,还有15个基因与α-DGP有关(POMT1、POMT2、POMGNT1、
FKTN、FKRP、LARGE、ISPD、POMGNT 2、DAG 1、TMEM 5、β 3 GALNT 2、POMK、β 4 GAT 1、GMPPB、LNPP5K)[9-10]。α-DGP主要表现为肌营养不良以及不同程度的中枢神经系统和眼部症状,可合并智力运动发育障碍。研究显示,DPM 3基因变异患者可以同时具有N-糖基化和O-甘露糖基化的表型特征[11];由DPM2基因变异所致CDG患儿可表现为发育迟缓、难治性癫痫、肌张力低下、进行性小头畸形及血清CK升高,肌肉活检示存在O-甘露糖基化异常[12]。此外,由DPM1基因变异所致的CDG-Ie患儿表现为小头畸形、肌张力下降、严重运动发育迟缓及CK升高,肌肉活检免疫组化染色示α-DG 糖基化水平降低,与α-DGP临床表型相符,同时证实DPM1基因变异可降低DPM转移酶活性[12]。以上多项研究表明,在临床上DPM复合物的任何亚基内的缺陷都可能导致N-糖基化和O-甘露糖基化障碍,O-甘露糖基化障碍可导致肌营养不良表型[6,11-13]。
CDG-Ie临床可表现为中枢神经系统、肌肉系统症状,特殊面容,眼部受累等。中枢神经系统表现包括运动、智力发育迟缓、小头畸形、脑萎缩、癫痫发作、肌张力低下、共济失调、步态失调等;累及肌肉可表现为肌力下降、血清肌酸肌酶升高等肌营养不良表型;特殊面容包括扁平鼻、高鹗弓、倒V 形嘴、鲤鱼形嘴、脚趾长、趾间间隙明显、关节挛缩等;眼部症状包括斜视、皮质盲、眼球震颤、视神经萎缩等;部分患儿可合并严重的胃肠道受累和凝血功能障碍。患者头颅MRI可表现为髓鞘发育延迟、脑萎缩、T2像上齿状核高信号等[14-18]。本例患儿存在智力、运动发育迟缓,小头畸形,癫痫性脑病,肌力、肌张力减弱,双足挛缩,扁鼻梁、小下颌特殊面容,双眼上斜,追光差;血清CK升高;头颅MRI 示脑萎缩,脑内髓鞘化明显偏弱。与既往报道CDG-Ie临床表现相符。一项对27例CDG合并早期癫痫性脑病患者的研究表明,CDG癫痫发作形式包括肌阵挛发作、阵挛发作和局灶性发作,进而进展为强直性阵挛性癫痫发作,婴儿痉挛;脑电图异常包括局灶性和多灶性癫痫放电,减慢的背景节律以及广泛的癫痫活动,暴发-抑制模式和癫痫持续状态[19]。此外,本例患儿存在癫痫性脑病,结合其癫痫发作起病年龄及发作形式,未考虑为大田原综合征。
DPM 1基因是CDG-Ie 的致病基因,本病为常染色体隐性遗传方式。本例患儿存在DPM1基因c.669-3C>G和c.677G>T(p.Arg226Leu)复合杂合变异,其中 c.677G>T变异导致第226号氨基酸由Arg(精氨酸)变为 Leu(亮氨酸)。上述两个变异均可能导致蛋白质功能受到影响。对其父母进行Sanger验证,母亲为c.669-3C>G变异携带者,父亲为c.677G>T变异携带者。以上变异不属于多态性变化,在人群中发生的频率极低。经查询human gene mutation database(HGMD)和PubMed等人类基因变异数据库,本例患儿DPM1基因变异位点为未曾报道的新发变异。因c.669-3C>G为内含子变异,故未进行基因软件预测其致病性;c.677G>T变异通过Polyphen-2、MutationTaster和SIFT三种基因软件预测,均为有可能致病。结合基因型、家族史、临床特点及相关文献报道,患儿最终诊断为CDG-Ie。
大多数CDG 以对症支持治疗为主,仅少数有潜在的治疗方法,包括膳食补充(例如,半乳糖用于PGM1-CDG,岩藻糖用于SLC35C1-CDG,锰制剂用于TMEM 165-CDG 或甘露糖用于MPI-CDG)和器官移植(例如,MPI-CDG 可进行肝脏移植,DOLK-CDG可进行心脏移植)[20]。由于CDG 涉及多器官系统损害,在临床管理上应针对患儿具体的表型和疾病的进程进行个体化管理,必要时需多学科联合。对症支持治疗包括给予高热卡的配方奶喂养,生长发育迟缓应及早进行物理治疗及康复训练,定期眼科随诊以诊断出潜在的眼部异常,并根据需要制定保护视力的措施(配镜、修补或手术)等。生酮饮食已证明可用于控制难治性癫痫发作。此外,遗传咨询对于CDG家庭尤为重要,随着遗传学分子技术的发展,对于有先证者的家庭,为其提供较为明确的遗传咨询及产前诊断,进行优生优育,以减轻患儿家庭及社会的负担。
综上所述,CDG-Ie是CDG中的罕见类型,目前报道的病例较少。患儿起病早,出生时或出生后数月即出现神经系统及肌营养不良表型,基因检测有助于明确诊断。
