短链酰基辅酶A 脱氢酶缺乏症患儿临床特点及基因变异分析
2020-09-25王伟青李文杰宋东坡于春冬吕金峰陈艳萍
王伟青 李文杰 宋东坡 于春冬 吕金峰 陈艳萍
青岛市妇女儿童医院新生儿筛查实验室(山东青岛 266000)
短链酰基辅酶A 脱氢酶(short-chain acyl-CoA dehydrogenase,SCAD)位于真核生物的线粒体中,可以降解短链脂肪酸,属于生物酰基辅酶A 脱氢酶(acyl-CoA dehydrogenase,ACAD)家族。1985年发现第1 例SCAD 缺乏症(SCAD deficiency,SCADD)。SCADD 是由于ACADS基因缺陷,导致短链酰基肉碱异常升高的一种常染色体隐性遗传代谢病。SCADD 临床表现多变,最常见的特征是新生儿发育迟缓,肌肉张力减退,喂养困难,癫痫,发育不良,发育畸形等。SCAD 活性受损导致血液中丁酰基肉碱(butyrylcarnitine,C4)和尿液中乙基丙二酸(ethylmalonic acid,EMA)浓度增加[1],可以作为SCADD诊断的重要标志物[2]。
1 临床资料
2015年1月到2018年12月青岛市共有476 379例活产新生儿,其中参加青岛市新生儿疾病筛查的有473 777例,筛查率为99.5%。筛查前所有家长均签署知情同意书。
筛查采用新生儿出生72 小时后的末梢血干滤纸片,以WATERS液相色谱-串联TQ Detector质谱仪,PerkinElmer公司的非衍生化多种氨基酸、肉碱测定试剂盒监测标本中的氨基酸及肉碱。尿有机酸以气相色谱-质谱联用分析仪分析。
共296 627例新生儿采用串联质谱技术进行氨基酸检测,初次筛查可疑阳性人数为4 864(1.6%),可疑SCADD 460例(0.16%)。经复查串联质谱短链酰基肉碱仍然升高的新生儿,行尿有机酸分析和基因二代测序。
收集新生儿和监护人的外周血,然后从外周血白细胞中提取基因组DNA。设计引物以ACADS的外显子和外显子-内含子边界为目标,通过聚合酶链式反应(polymerase chain reaction,PCR)扩增,PCR产物通过Sanger直接测序进行分析。最终确诊SCADD患儿7例,发病率为1/42 375。
7 例SCADD 患儿均为足月儿,体格检查未见异常,四肢肌张力可,原始反射可引出。早期诊断确诊后开始饮食治疗,避免饥饿及感染,避免低血糖,低脂饮食。7例患儿均无临床症状,因此未服用左旋肉碱或维生素B2等药物。跟踪随访至少3个月,所有患儿神经系统发育均无异常,没有骨骼肌张力减退或张力过高,生长良好,发育评估无异常。血清生化指标无异常,见表1。
基因检测结果显示,确诊患儿存在ACADS基因变异,分别为c.1029+89_1029+90insC,c.1031A>G,c.1130C>T,c.1157G>A,c.1186G>A,c.164C>T,c.445A>T,c.949A>G,c.989G>A。其中c.1031A>G(HGMD CM085212),c.1157G>A(rs766183395),c.164 C>T(HGMD CM 085955,rs 147442301),c.989 G>A(HGMD CM 067634)为致病变异,与SCADD发生有关,可能导致剪接部位变化,氨基酸序列改变,蛋白质特性可能受到影响。ACADS基因变异c.1130C>T,c.1186G>A,c.445A>T,c.949A>G未见文献报道,为临床意义未明变异。
phastCons和phyloP可用于确定核苷酸保守程度,phastCons值在0到1之间变化,基于46种不同物种的基因组序列的多重比对反映每个核苷酸属于保守元件的可能性。phyloP/phastCons值越接近1,核苷酸越可能是保守的,变异有害的可能性越大。c.1130C>T,c.1186G>A,c.445A>T,c.949A>G变异的phyloP/phastCons值分别为0.998、1.000、0.999、1.000,基因位点高度保守,变异有害的可能性较大。使用PROVEAN和SIFT软件对变异位点进行风险评估,结果均为有害(-9.11、-3.91、-3.55、-3.70)/(0.000、0.001、0.022、0.001)
6 例患儿的ACADS基因存在复合杂合变异,1 例患儿存在c.1031A>G纯合变异,变异位点均分别来自父母。如例2 患儿ACADS基因的第10 号外显子上新发的母本等位基因错义变异c.1186G > A(p.E396K),与第9 外显子内含子连接处的父本等位基因变异c.1029+89_1029+90 ins C(SNP,rs397744635),导致第396位的谷氨酸变为赖氨酸,编码区第1029位碱基后89 与90 内含子间插入了一个胞嘧啶,改变了氨基酸序列,影响蛋白质结构与功能,最终导致SCAD功能异常或合成障碍,脂肪酸β 氧化受阻。目前在dbSNP数据库(Genomes Project)中已有c.1029 +89_1029+1029+90 insC等变异的报道,但没有更多致病机制的研究信息。
2 讨论
新英格兰的SCADD的新生儿发病率为1/33000,加利福尼亚约为1/34632[3],美国各地区SCADD 的患病率在1/330000~3/33000之间[4],中国尚未有SCADD患病率的准确数据,对SCADD患者进行流行病学调查是很有必要的。
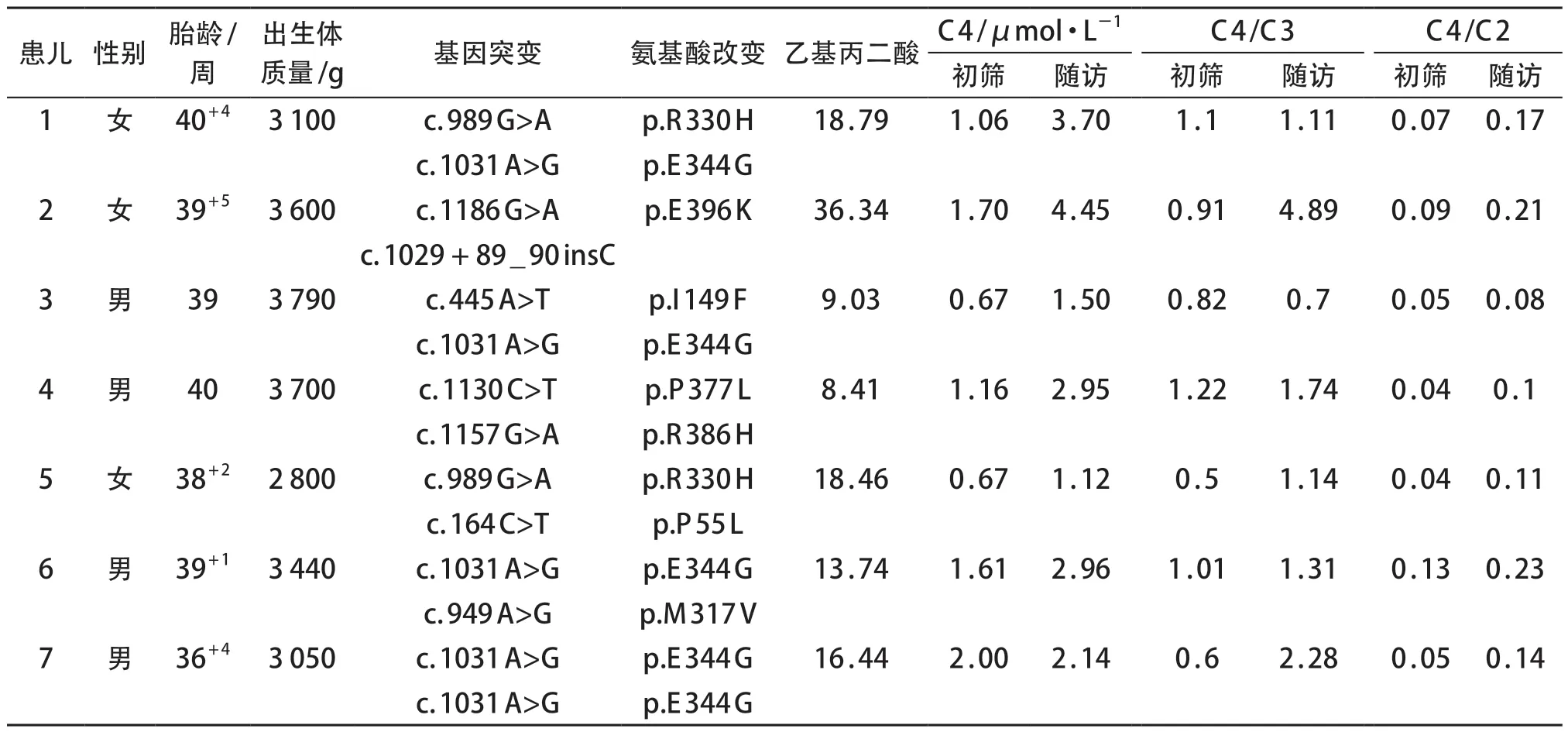
表1 7例SCADD患儿基本情况及临床指标
ACADS是SCAD的致病基因,PubMed已记录162种变异。目前报道多例有症状的SCADD患者,大部分是复合杂合变异或纯合变异。大多数SCADD 患者的初始症状表现为低血糖或神经症状。ACADS 变异与SCADD的临床症状和病情严重程度的关系尚不明确,纯合变异可能与临床疾病的易感性有关,在压力或饥饿等应激条件下,β 氧化能量供应不足,携带变异的患者可能诱发SCADD。
研究者已克隆并测序了编码人胎盘SCAD前体的cDNA。编码SCAD的ACADS基因位于染色体12q24上,跨越14.2kb的DNA,其中1.9 kb的编码区分裂成10个外显子,编码412个氨基酸,其中包括24个氨基酸的前导肽。与其他酰基辅酶A脱氢酶基因家族成员一样,SCAD 是四聚体线粒体黄素蛋白,它以前体形式转运到线粒体中,并经过蛋白水解加工成成熟形式发挥作用。核基因组编码SCAD,是脂肪酸氧化系统中的线粒体酶,催化β氧化螺旋的第一步,将四或六个碳链的酰基辅酶A转化为2-烯酰辅酶A,介导丁酰辅酶A的脱氢反应,并将电子传递给电子传递黄素蛋白(electron transport flavoprotein,ETE)[5]。丁酰基肉碱是SCAD的主要底物,SCADD患者尿液、血液和细胞中的丁酰基肉碱,丁酰甘氨酸,EMA和甲基琥珀酸等代谢物升高。研究证实,有SNP(rs2055938的G等位基因)的儿童或成人可能与血清C 4 水平升高有关,但两者的相关性无法确定,这也是导致新生儿C4浓度存在25%变异的原因[6]。有或者无明显症状的SCADD患者C4均升高,可以通过新生儿遗传代谢病筛查并进一步通过基因检测确诊。
随着20 世纪90 年代基因测序诊断方法的发展,有望确定脂肪酸氧化缺陷基因型与表型之间的关系,从而通过预测基因变异的结果指导临床治疗。研究证明,无义变异、剪接点改变和移码变异可以影响无义介导的mRNA衰变(nonsense-mediated mRNA decay,NMD)系统,导致mRNA 降解,进一步表现为严重的临床症状,而微小的插入和缺失变异以及错义变异可能与较温和的临床症状有关[7]。但SCADD 患者的代谢情况,酶浓度不能通过基因变异来预测,SCAD 功能障碍程度与临床表现之间没有明确关系,严重的ACADS变异可能并不会有严重的临床表现。
大多数SCADD患者携带纯合变异或复合杂合变异,或ACADS基因伴失活变异。常见ACADS变异是错义变异,在欧洲人群中5和6号外显子中c.511C> T和c.625 G> A变异最常见,而美国人群中c.511C> T的等位基因频率为0.3%,c.625G> A的等位基因频率为5.5%[8-9]。几乎所有SCADD患者的ACADS中都包含错义变异,通过改变折叠方式和细胞的寿命来影响蛋白质的生物合成[10]。
SCAD 缺陷小鼠与野生型小鼠相比,蛋白错误折叠的趋势和异常蛋白的浓度增加,这种现象表明变异可能是功能获得性细胞表型[11]。c.1130 C>T,c.1186 G>A,c.445 A>T,c.949 A>G 被证实为是新的变异,在PubMed、dbSNP数据库、1000 Genomes Project数据库或Human Gene Mutation Database中均不存在。这些变异可以改变氨基酸序列,从而改变蛋白质的疏水性,例如10号外显子中的C.1186G>A使396位的谷氨酸(Glu)变为赖氨酸(Lys)(p.E396K),谷氨酸的疏水性从-3.5变为-3.9[12]。为了阻止与其他线粒体成分的疏水相互作用,某些易受攻击的SCAD 蛋白被折叠,这种突变通过改变氨基酸的疏水性来改变蛋白折叠方式。作为基质蛋白,SCAD 在线粒体hsp70的协助下被转移到基质中,然后在线粒体hsp70和hsp60/10的协助下折叠成功能结构,并定位到活性位置[13]。在基质之前折叠方式是正确的,小部分具有错义变异的蛋白质可能错误折叠成只具有部分功能的结构,而其余的错误折叠可能在体内积累或直接降解,无法达到完全折叠的功能蛋白的构象。折叠中间体的降解导致功能丧失。如果降解速率较低,并且依赖于基因与环境的相互作用,错误折叠的蛋白质可能会积累和干扰正常的细胞功能,这可能会与其他蛋白质的产生相互作用[14]。由ACADS变异引起的SCAD 蛋白错误折叠可能会改变其催化特性和结构,从而赋予疾病易感性[15],识别这些促成因素有助于有效诊断和治疗SCADD。
本研究中7 例SCADD 患儿均为新生儿筛查早期确诊,确诊后采取低脂饮食治疗,未服用药物。虽然通过软件预测大多数变异为有害变异,但除例3患儿失访外,其他患儿均没有出现SCADD的临床表现,如喂养困难、发育迟缓、肌张力减退、癫痫等,仅有血液中C4水平持续偏高,推测ACADS基因变异与血清C4水平有相关性,但与临床症状相关性不高。在早期确诊后经饮食干预可以避免新生儿面临饥饿、压力等应激状态,减少SCADD发病的可能性。
综上,新生儿疾病筛查可以早期诊断、治疗SCADD,延缓发病改善预后,对新生儿健康是重大的公共卫生举措。
