内毒素对大鼠心肌肌质网功能的抑制作用
2020-09-02陈怀生刘雪燕郭晓静洪澄英李威曹静
陈怀生,刘雪燕*,郭晓静,洪澄英,李威,曹静
1南方科技大学第一附属医院/暨南大学第二临床医学院/深圳市人民医院重症医学科,广东深圳 518020;2南方科技大学第一附属医院/暨南大学第二临床医学院/深圳市人民医院病理科,广东深圳 518020
脓毒症是严重感染导致的致命性多脏器功能障碍综合征[1],病死率可达20%~30%[2]。大约60%的脓毒症患者伴有心功能不全[3-5],这类患者的病死率更高[4]。革兰阴性杆菌感染是引起脓毒症的主要原因。内毒素即脂多糖(lipopolysaccharide,LPS),是革兰阴性细菌细胞壁的成分,在脓毒症、多脏器衰竭(multiple organ failure,MOF)的发生发展中起关键作用[6]。LPS通过与白细胞、巨噬细胞Toll样受体(Toll-like receptor,TLR)2、4结合,调节TLR信号转导通路,影响患者的免疫功能,从而引起全身炎症反应综合征[7]。研究发现,心肌细胞表面可能表达LPS受体,或促进TLR受体生成,通过损伤相关分子模式(damage associated molecular patterns,DAMP)诱导信号转导,最终影响心肌细胞线粒体[5]和肌质网功能,导致心功能不全。本研究拟通过观察内毒素致大鼠心肌肌质网相关酶学改变情况,评价内毒素对大鼠心肌肌质网功能的抑制作用。
1 材料与方法
1.1 主要试剂与设备 脂多糖(LPS fromEscherichia coliD55:B5)购自美国Sigma公司。无创心率计、血压计购自上海宇扬仪器公司;透射电子显微镜(Philips EM208s)购自荷兰Philips公司。
1.2 脓毒症大鼠模型的建立与分组 健康成年雄性SD大鼠10只,体重180~220 g,由华大基因公司提供。随机分为空白对照组与内毒素注射组,每组5只。内毒素注射组从大鼠尾静脉注射脂多糖(1 mg/ml,以生理盐水为溶剂,按0.7 mg/kg给药),1次/d,连续2 d,建立脓毒症大鼠模型;空白对照组不做处理。本实验过程符合国家和单位有关实验动物管理和使用的规定。
1.3 大鼠血流动力学参数监测 每日于实验大鼠尾静脉注射脂多糖2 h后,采用无创心率计和血压计记录两组大鼠的心率(次/min)、平均动脉血压(mean atrial pressure,MAP,mmHg)。
1.4 大鼠心肌组织及细胞病理形态学评价 第二次给予脂多糖4 h后,腹腔注射10%水合氯醛(0.3 ml/100 g)处死动物。取部分心肌组织,采用多聚甲醛固定,石蜡包埋、切片,HE染色后光镜下观察其病理结构变化。另取部分心肌组织,经戊二醛-多聚甲醛和锇酸-亚铁氰化钾固定,乙醇、丙酮依次脱水,用乙氧基树脂浸透包埋,然后制备电镜切片,切片经醋酸铀、柠檬酸铅染色,在透射电子显微镜下观察心肌线粒体超微结构。
1.5 大鼠心室肌细胞肌质网钙调节蛋白mRNA水平检测 提取大鼠心室肌总RNA,以GAPDH为内参,采用反转录实时定量PCR(reverse transcription-qPCR,RT-qPCR)检测肌质网肌钙集蛋白(calsequestrin,CASQ1)、钠-钙交换体(Na+-Ca2+exchanger, NCX)、兰尼碱受体2型(ryanodine receptor,RyR2)、钙调蛋白磷酸酶1(protein phosphatase type 1α,ppplCa)、肌质网Ca2+-ATP酶(sarcoplasmic reticulum Ca2+ATPase,SERCA2)、受磷蛋白(phospholamban,PLN)、ADP/ATP移位酶(mitochondrial carrier,adenine nucleotide translocator 25,member 4,slc25a4)的mRNA表达水平。引物序列见表1。PCR反应条件:94 ℃预变性10 s;94 ℃5 s,60 ℃ 35 s,40个循环。采用2-ΔΔCt法计算mRNA相对表达量。

表1 大鼠心肌肌质网钙调节蛋白的引物序列Tab.1 The primer sequences of myocardial sarcoplasmic reticulum Ca2+ handling proteins in rats
1.6 统计学处理 采用SPSS 19.0软件进行统计分析。计量资料以表示,组间比较采用t检验,P<0.05为差异有统计学意义。
2 结 果
2.1 大鼠血流动力学参数变化 注射首日及次日内毒素注射组大鼠的心率与空白对照组比较均明显升高,差异有统计学意义(P<0.01)。内毒素注射组大鼠注射首日MAP与空白对照组比较明显降低(P<0.05);注射次日两组MAP差异无统计学意义(P>0.05,图1)。
2.2 大鼠心肌组织病理形态学改变 空白对照组大鼠心肌组织致密,未见明显血管扩张,细胞排列整齐;电子显微镜下肌纤维排列有序,线粒体、肌质网规则可见。内毒素注射组大鼠心肌细胞排列疏松,细胞间可见炎性细胞浸润;电镜下心肌肌纤维断裂,心肌细胞线粒体、肌质网形态辨认困难(图2)。

图1 内毒素对大鼠心率及MAP的影响Fig.1 Effects of lipopolysaccharide on heart rate and mean arterial pressure of rats
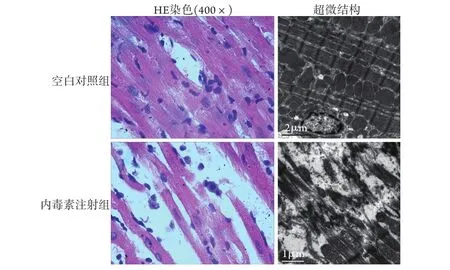
图2 内毒素致大鼠心肌组织病理形态学的改变Fig.2 Changes of myocardial histopathology induced by lipopolysaccharide in rats
2.3 大鼠心室肌细胞肌质网钙调节蛋白mRNA表达情况 RT-qPCR检测结果显示,注射脂多糖后,大鼠心室肌CASQ1、NCX、ppplCa、PLN、SERCA2 mRNA表达水平与空白对照组比较均明显增加(P<0.05),而两组RyR2、slc25a4 mRNA表达水平差异无统计学意义(P>0.05,图3)。

图3 大鼠心室肌细胞肌质网钙调节蛋白mRNA表达情况Fig.3 mRNA levels of sarcoplasmic reticulum Ca2+ handling proteins in ventricular myocytes of rats
3 讨 论
随着心脏超声在ICU的广泛应用,脓毒症合并心功能不全更容易被发现[8]。超声显示,严重脓毒症患者常合并以左室射血分数(left ventricular ejective factor,LVEF)下降、心室扩张为特征的心室功能减退[9]。鉴于引起脓毒症的主要原因是革兰阴性菌感染,本研究通过静脉注射脂多糖建立脓毒症大鼠模型,发现脓毒症大鼠心率明显增快,MAP降低;并可见较明显的心肌损伤,其心肌细胞排列紊乱,细胞间出现炎性细胞浸润,电子显微镜下可见明显的肌纤维断裂,线粒体结构受到不同程度的破坏。RT-qPCR检测发现,脓毒症大鼠出现心肌细胞肌质网酶学异常变化,CASQ1、NCX、ppplCa、PLN、SERCA2等酶的mRNA水平较空白对照组升高。由此可见,脓毒症心肌病的机制较为复杂,结合以往研究总结如下。
首先,内毒素和炎性因子直接损伤脓毒症机体心肌细胞。LPS可直接作用于心肌细胞LPS受体,通过信号转导作用影响心肌收缩力;还可促进心肌细胞TLR的表达[10]。LPS诱导的TLR以TLR4为主,可导致心肌细胞IL-1β和TNF-α mRNA表达上调[11],血清IL-1β、TNF-α浓度增加,继而引起心肌线粒体功能障碍。心肌TLR4表达缺乏的动物对LPS反应减弱,导致LPS对心肌功能影响较小[12];敲除TLR4基因的大鼠注射LPS后,可观察到心脏功能障碍弱于野生型[11]。另外,TNF-α可能与心肌细胞膜的特异性受体结合,促进内质网释放过多钙离子,使心肌线粒体钙离子内流增加[13-14]、积聚、超负荷,导致线粒体肿胀、破坏。TNF-α还可启动心肌线粒体凋亡信号通路,使线粒体通透性转换孔呈高通透状态,H+内流,导致线粒体膜电位缺失,线粒体肿胀、破裂。
脓毒症患者常出现心肌舒张功能障碍。正常状态下心肌舒张时,心肌细胞通过SERCA2摄取胞质内的钙离子进入肌质网,PLN磷酸化可促进SERCA2对钙离子的摄取。在这个过程中,2磷蛋白(2 phosphoproteins)、蛋白磷酸酶1抑制物(inhibitor-1 of protein phosphatise 1)和小热休克蛋白20(small heat shock protein 20)也可发挥调节作用[15]。脓毒症状态下,SERCA2a可出现代偿性的活化增强,这种异常活化导致脓毒症动物的心脏舒张功能障碍[16],某些PLN磷酸化阶段的类似物则能够影响SERCA功能[17]。
其次,细菌毒素和细胞因子对心功能具有间接抑制作用。脓毒症患者TNF-α增高与心肌收缩和舒张功能障碍呈正相关,这一作用与TNF-α诱导一氧化氮合酶(nitric oxide synthetase,NOS)表达增加,导致心肌细胞NO浓度增高有关[18]。脓毒症时这些细胞因子介导NOS类似物表达异常增加,进而催化L-精氨酸合成增多,导致心肌细胞NO合成增多并失调[19]。NO对心肌细胞相关基因具有毒性效应,可升高环磷酸鸟苷的水平,引起Ca2+内流减少,抑制了心肌收缩功能[20];同时可导致线粒体复合体Ⅰ和复合体Ⅱ受抑,影响线粒体的呼吸功能和能量代谢,直接损伤心肌细胞[21];NO还可导致血管过度扩张,其引起的低血压加剧了心肌的低灌注。
再者,氧化应激反应在脓毒症心肌抑制中扮演着重要角色。由于炎症介质和炎症因子的介导,氧自由基(oxygen free radical,OFR)产生增多。OFR包括超氧阴离子自由基(>)和羟自由基(.OH),在心肌组织超氧化物歧化酶的作用下,生成过氧化氢,后者可被过氧化氢酶或谷胱甘肽过氧化物酶(GSH-PX)灭活。脓毒症时由于灭活机制异常,活性氧大量产生、积聚,清除困难,表现为丙二醛(malondialdehyde,MDA)增高、超氧化物岐化酶(superoxide dismutase,SOD)降低,从而影响心肌线粒体ATP的产生,导致细胞发生致命的能量衰竭[22]。
最后,脓毒症可导致心肌细胞凋亡。发生脓毒症时,TNF可通过内、外源性途径诱导细胞凋亡的发生,内源性途径主要由caspase-8介导,外源性途径则由caspase-9介导,两个途径共同活化凋亡酶caspase-3及caspase瀑布,导致心肌细胞及其他细胞的凋亡[23]。
综上所述,内毒素可能通过多种机制,尤其是引起肌质网钙调节蛋白相关酶SERCA2a、CASQ1、NCX、ppplCa、PLN等的过度活化,导致肌质网钙调节异常,从而抑制心肌细胞舒张功能,最终收缩功能也受到影响。
