光催化环己烷选择氧化合成环己酮/环己醇催化研究进展
2020-08-17佟占鑫石亮彭超李成宇秦弘宇陈丽娟向育君
佟占鑫,石亮,彭超,李成宇,秦弘宇,陈丽娟,2,向育君
(1 湖南科技大学材料科学与工程学院,湖南湘潭411201;2 湖南省能量转换与储能材料重点实验室,湖南湘潭411201;3 湖南科技大学生命科学学院,湖南湘潭411201)
环己烷氧化产物环己醇和环己酮是己内酰胺合成中的重要中间体,而己内酰胺是尼龙生产中的重要原料[1-3]。热力学上,由于环己烷分子饱和C—H键活化能高(97kcal/mol),环己烷选择性氧化需在高温条件下进行(O2压强为1.5MPa,150℃),极易产生副产物,环己烷转化率需控制在4%以下(以获得约80%的环己醇和环己酮选择性)[4]。因此亟需更温和、更环保的反应路线来取代这一高能耗、选择性低的反应路径。近年来,以太阳光为驱动力的光催化技术,特别是可见光响应的光催化材料研究取得了很大进展,开发高效选择性可见光响应光催化剂,实现温和条件下环己烷光催化氧化有望成为一条更理想的环己烷选择性氧化途径[5-6]。本文总结了近十年来光催化环己烷选择性氧化的研究进展,并分别从光催化体系、反应机理和反应影响因素等方面进行讨论。
1 光催化环己烷选择性氧化催化体系及反应机理
环己烷光催化氧化常用的催化剂主要有氧化物半导体光催化剂、非金属氧酸盐光催化剂、有机及非金属光催化剂、表面等离子体光催化剂等。
1.1 氧化物半导体光催化剂及机理
环己烷光催化氧化的氧化物半导体光催化剂包括TiO2基催化剂和非TiO2基催化剂。Moulijin 研究组[7-14]对TiO2催化剂进行了系统研究,以原位傅里叶变换衰减全反射红外光谱法(ATR-FTIR)技术、同位素标记技术等对不同商业级TiO2光催化环己烷选择性氧化从晶体结构、形貌粒径、表面酸性、入射光通量和波长范围、水蒸气的影响等方面进行了系统的深入研究,探索了表面活性自由基形成及其作用机理、产物组成及影响因素、失活原因及其影响因素。研究结果表明TiO2光催化环己烷选择性氧化反应是基于Mars-van Krevelen 机理(图1),环己烷由表面TiIV-OH 物种活化形成环己基自由基,环己基自由基经双TiIVOH 物种协同氧化生成环己酮(图1,途径1)或和O2、TiIV-OH 物种协同氧化生成环己酮(图1,途径2)。
由于TiO2的禁带能隙宽,需要紫外光激发,而紫外光激发下TiO2的氧化能力强,容易引发副反应。因此,近年来可见光激发的改性TiO2催化剂获得重要关注。表1从反应条件、转化率和选择性等方面对部分改性TiO2光催化剂如FeO@TLO(层状钛 酸 盐)[15]、 V2O5@TiO2[16]、 NH2-MIL-125(Ti)/TiO2[17]、TiO2/rGO[18]进行了综合比较。
在这些改性TiO2催化剂中,催化剂的改性或复合成分的作用包括:修饰TiO2的能带结构、作为电子“储库”以促进光生电子转移[16,18]、调变TiO2表面性质和表面中心浓度、提供氧化还原中心[15]。如在FeO@TiO2催化剂中,FeOx中心取代部分TiO2的表面羟基,促进部分氧化产物环己酮和环己醇的脱附,同时FeOx中心可接受TiO2激发产生的光电子,促进电子-空穴分离[15]。在V2O5@TiO2中,在光激发下,V 中心能结合光生电子,然后传递电子给O2,在水分子共同作用下生成·OOH,并和环己烷分子反应生成C6H11OOH,分解成环己酮/醇[16]。在TiO2/还原氧化石墨烯(TiO2/rGO)中,rGO 接收TiO2激发的光电子,促进电子空穴的有效分离,同时rGO 能调变催化剂氧化性能,使O2按双电子转移机理还原活化,抑制O2的单电子还原物种O2-生成,提高环己酮的选择性[18]。

表1 改性TiO2光催化环己烷选择性氧化实验结果

图1 TiO2光催化环己烷氧化中环己酮生成过程[3]
其他氧化物光催化剂还包括表面憎水修饰Cr-SiO2[19]、Cr/Si 混 合 氧 化 物[20]、V2O5@Al2O3[21-22]、Cr-Si 二元氧化物[23]、Cr/Ti/Si 三元氧化物[24]、硫化钒掺杂中空TS-1[25]、C-N 掺杂Cr2O3[26]、FexOy/Ti 修饰介孔SiO2[27]、Ti-HUS-2[28]等。Tanaka 等[21]研 究了V2O5@Al2O3催化的环己烷光催化选择性氧化反应,指出保持较高的气相O2浓度和辐射光波长大于330nm 是获得较高氧化产物选择性的关键,优化条件下环己酮选择性为87%,酮/醇比为3.8。Shiraishi 等[23-24]研究了溶胶-凝胶法制备的Cr-Si 二元氧化物Cr-SiO2和Cr/Ti/Si 三元氧化物Cr0.04Ti0.04Si的光催化环己烷选择性氧化反应,并和浸渍法制备的Cr/SiO2和模板法制备的Cr&MCM-41 进行比较。在二元和三元氧化物催化剂中,低氧化激发态Cr4+(Td4+*)为反应的活性物种,由端基氧相连的六配位Cr6+(Td6+)在光激发还原下转变成。在Cr/SiO2或Cr&MCM-41 中,Si 在Cr6+中心的分布为“伞形”(umbonal)结构,Si 和Cr6+中心的端O 原子较远。在Cr-Si 二元氧化物中,Si 在Cr6+中心的分布为“均相”(homogeneous)结构,端O 原子和附近Si 中心容易成键,电荷部分转移至Si 原子上,使Cr6+更易还原成Td4+*物种(图2)[23]。Cr/Ti/Si 三元氧化物活性比Cr-Si 二元氧化物更高,是因为Cr 和Ti 间形成Cr—O—Si 键,其离域作用使光激发下产生的Td4+*活性物种的电子稳定,抑制了Td4+*的失活,从而提高了光催化活性(图3)[24]。Mao 等[25]研究了硫化钒掺杂中空TS-1 等氧化物催化剂的光催化环己烷氧化性能,S、V 掺杂TS-1 中形成了O==V—O4—Si 桥连中心,桥键中的O==V 和O4—Ti 是环己烷光催化氧化的活性中心,添加浓HCl 和H2O 对光催化活性有明显的促进作用,这是因为HCl 和H2O 在光激发下产生Cl·和·OH,使环己烷脱氢活化产生环己基自由基。在Ti-HUS-2 催化剂中,通过嫁接制备法使TiV-acac 配合物进入层状HUS-2 的层间,然后煅烧使四配位Ti 以Si—O—Ti 键均匀分布在SiO2层中,获得比Ti-Si 分子筛TS-1 更高的活性[28]。
1.2 多金属氧酸盐光催化剂及机理
钒取代多金属氧酸盐催化剂也被用于光催化环己烷氧化反应[29-30],Fu 等[30]报道了XPMo12-nVnO40-HCl催化的光催化环己烷选择性氧化反应。研究表明该体系中多金属氧酸盐的VV-O-M 中心能捕集HCl,产生质子化光活化物种[PA, POM-(VVOHM)+Cl—],而溶剂乙腈也因弱配位作用参与PA的形成,反应机理如图4所示,多金属氧酸盐中V成分的增加能促进PA生成,反应体系中添加少量水,能捕获光激发产生的Cl·生成HClO,减少氯取代副产物形成[30]。

图2 Cr-Si二元氧化物光催化机理[23]
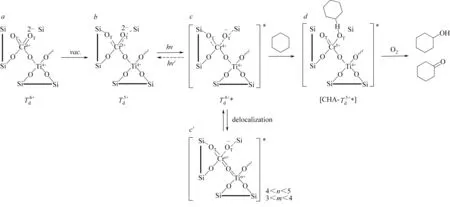
图3 Cr/Ti/Si三元氧化物光催化环己烷选择性氧化机理[24]
Liu等[31]报道了Ag3PW12O40/C3N4催化的光催化环己烷选择性氧化反应。在60℃,可见光辐射下,环己烷转化率为8.62%,并以99%的选择性生成环己酮,催化剂中C3N4在光激发下能使水氧化转变成H2O2,为反应的氧化剂,而Ag3PW12O40则是促进H2O2分解产生活性自由基HO·,使环己烷活化。
1.3 有机及非金属光催化剂及机理
有机化合物和非金属光催化体系报道不多,主要因为具有饱和C—H键的环己烷活化能高,反应性低,有机化合物和非金属光催化体系的光催化氧化活性较低,难以使环己烷分子活化而引发光催化反应。如9-均三甲基-10-甲基吖啶离子(Acr+-Mes)是一种重要的有机光催化剂,用于光催化氧化时,底物的单电子氧化电位需比9-均三甲基-10-甲基吖啶离子中的异丙叉丙酮基(Mes)单电子氧化电位低才能使底物分子活化[32-33],因此单独Acr+-Mes 不能用于环己烷光催化氧化,需要添加助催化剂。Fukuzumi 等[34]报道了由Acr+-Mes 和HCl组成的环己烷选择性氧化光催化体系,得到环己酮、环己醇和过氧化氢组成的产物。Cl-先和均三甲苯基自由基阳离子通过静电作用结合活性中间物种,在光激发下产生活性自由基Cl·,使环己烷活化。产物组成中,环己醇选择性为31%,环己酮选择性为50%[34]。
Shimizu等[35]研究了Cu-Y/H2O2在光激发下的环己烷选择性氧化,真正的催化剂是Y型分子筛上的Cu2+和H2O2形成3 种二氧桥铜配合物,在环己烷(10mmol)/H2O2(10mmol)/CuY (0.01g)、300K、光反应6h 条件下,产物组成中,环己醇选择性为54%,环己基过氧化氢选择性为37%,环己酮选择性为8.2%,CO2选择性为1.3%。反应机理如图5所示,在光(λ>340nm)激发下,二氧桥铜配合物分解并和环己烷分子反应生成环己基过氧化物,再转变成环己醇和环己酮产物[35]。
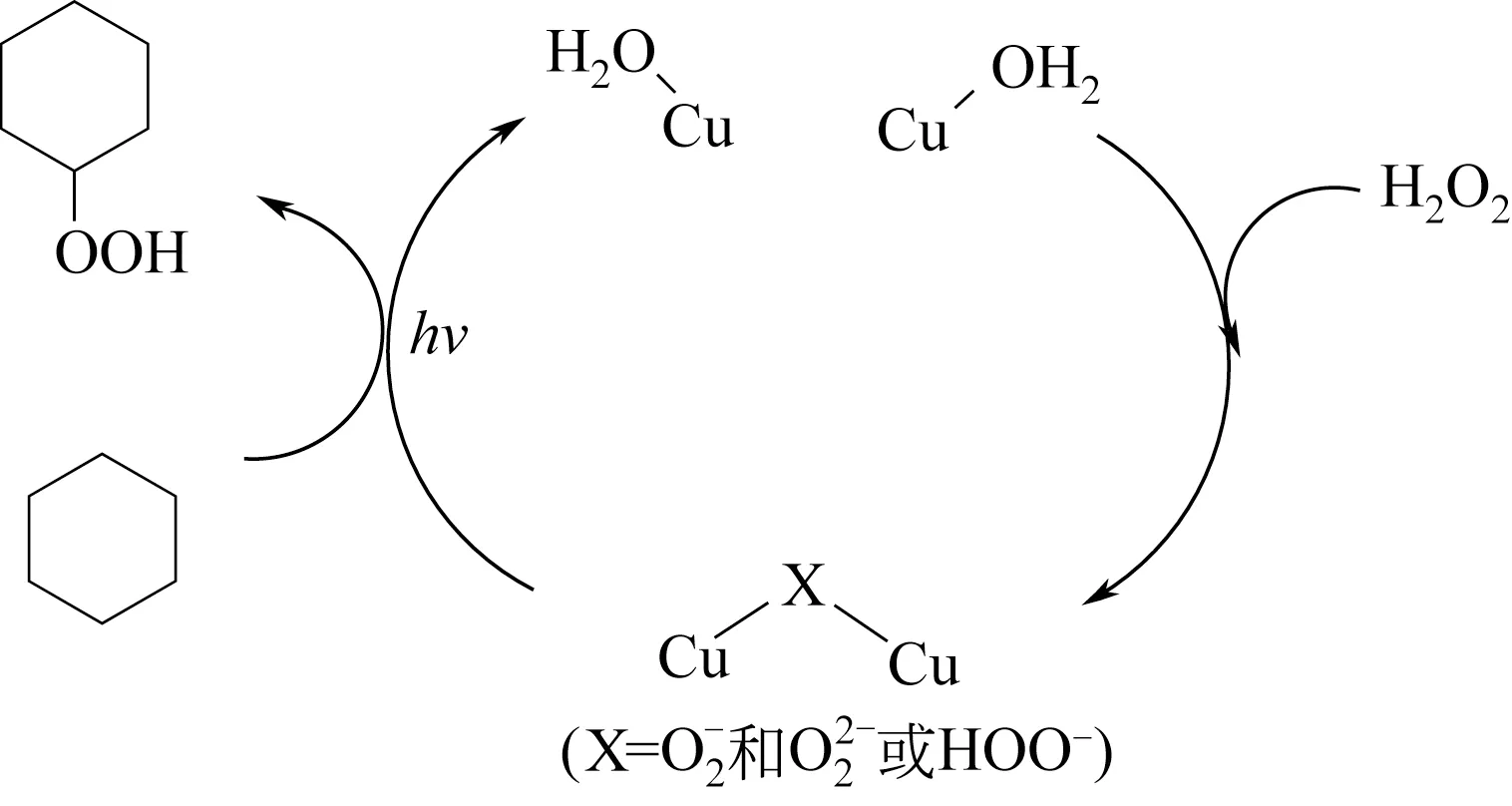
图5 Cu-Y/H2O2光催化环己烷选择性氧化反应机理[35]

1.4 表面等离子体光催化剂及机理
半导体光催化剂的一个主要弊端是光生载流子易于复合,其原因是由于光子穿透深度超过空间电荷区宽度,而光生载流子的平均自由程短,在到达催化剂表面参与光化学反应前容易复合湮灭[37-39]。复合其他半导体在界面内建电场作用下或在半导体晶体掺杂下引入杂质能级可促进光生电荷有效分离,但容易引起晶体较大畸变。而在不导致半导体晶体结构产生较大畸变下,在半导体表面沉积具有表面等离子共振效应(SPR)的金属纳米粒子是改善半导体光催化性能的有效途径[40]。SPR是指金属中导带自由电子在和电磁场作用引发的集体振荡运动,沉积在半导体表面的金属纳米粒子通过SPR效应会增加可见光激发下的局域场能,促使更多激发态电子参与光催化氧化还原反应和能量转换过程[41-42]。SPR金属纳米粒子/半导体复合催化剂的光催化性能依赖于金属纳米粒子和半导体的功函数、金属纳米粒子的尺寸和形貌等因素。金属纳米粒子的表面沉积会改善半导体在可见光区域的光吸收特性。因此,在光催化环己烷氧化中,表面等离子体光催化剂主要用于可见光激发的环己烷选择性氧化反应[43-54]。
Liu 等[43]报道了氧化石墨烯/银纳米棒复合光催化剂,比较了纳米银形貌(粒子和棒状)、尺寸的影响。在室温和模拟太阳光条件下,以叔丁基过氧化氢(TBHP)为氧化剂,反应48h,以氧化石墨烯/银纳米棒(平均粒径10nm,长径比9~12)为催化剂,环己烷转化率最高达37%,环己酮/醇总选择性达94.0%。最近,这一课题组报道了由Au-WO3催化的光热协同作用下环己烷选择性氧化反应,以质量分数为2%的Au-WO3为催化剂、在1.5MPa 干燥空气压力、模拟太阳光Xe 灯光源、反应温度120℃条件下,反应8h,环己烷转化率为9%,环己酮/醇总选择性为99%[44]。采用光源没有加热和黑暗加热反应条件下,环己烷转化率分别为0.49%和7.61%[44]。对Au-WO3催化剂来说,光热协同作用下,热氧化反应为主导,但环己烷9%转化率比单加热与单光催化的环己烷转化率的总和(0.49%+7.61%)更高,说明光热两种引发方式具有协同促进效应[44]。
Liu 等[45]则报道了纳米金负载于碳纳米管内的Au-in-CNTs 催化剂的光催化环己烷选择性氧化反应,在模拟Xe 灯光源、TBHP 为引发剂、333K 反应条件下,反应48h 后,Au-in-CNTS 显示出最佳反应活性,环己烷转化率达14.64%,环己醇选择性达86.88%。作为对比的纳米金负载于碳纳米管外的Au-out-CNTs,活性则明显较低,环己烷转化率仅为3.45%,环己醇选择性为70.28%[45]。其活性的差异是由于负载于CNTs 孔内纳米金尺寸更小以及CNTs 孔结构的限域作用增加了催化剂活性位和反应物的接触时间所致。反应机理是纳米金吸收可见光,产生激发电子转移至碳纳米管,在纳米金/碳纳米管界面使O2通过二电子氧化还原反应(ORR)过程还原成H2O2,然后在TBHP 引发下分解成·OH,使环己烷转变成环己醇[45]。
除了贵金属纳米粒子外,Huang等[46]报道了Cu纳米粒子/C 量子点复合光催化剂,在60℃、Xe 灯模拟可见光源、TBHP 为氧化剂条件下,反应48h后,环己烷转化率为50.2%,环己酮选择性为78.3%,而单独以Cu 纳米粒子和C 量子点为催化剂,同等条件下,环己烷转化率仅为6.5%和8.1%。Cu 纳米粒子/C 量子点复合光催化剂的光催化性能还和入射光波长及强度密切相关,在和Cu纳米粒子表面等离子共振波长(540~640nm)匹配的红光辐射以及光强为60mW/cm2以上,环己烷转化率最高[46]。
总之,从不同催化体系的反应活性和选择性比较,一些光催化剂展示了很好的活性和选择性,其中V、Cr、Si修饰催化剂以及金属纳米粒子/半导体复合催化剂等,在可见光作用下环己烷转化率能达10%左右,选择性90%以上,优于工业环己烷氧化结果(转化率4%,选择性80%),HCl在某些催化体系中有很强的促进作用。光催化环己烷氧化产物中环己酮/环己醇比例(3~4)大于热氧化条件下环己酮/环己醇比(1~2),表现出不同的反应机理。目前光催化环己烷选择性氧化还主要停留在实验室阶段,进入工业应用还需进一步提高催化剂对全太阳光谱的响应性、光化学稳定性,设计工业光催化合成装置。
1.5 反应机理研究总结
环己烷的光催化选择性氧化机理包括:环己烷活化、选择性氧化产物环己酮/醇生成、活性自由基再生和催化剂失活机理。目前普遍认为,光辐射下,环己烷分子氧化脱氢形成环己基自由基是催化循环的起点,但形成环己基自由基的途径则有不同的观点:①直接被光生空穴氧化生成自由基,C6H12+h+→C6H11·[55];②催化剂表面官能团和光生空穴作用活化,再和环己烷反应生成自由基,如Ti-OH+h+→Ti-OH·,C6H12+(OH·)ads→C6H11·[8];③由氧化剂如TBHP或O2的光分解或还原产生活性自由基OH·,使环己烷转变成环己基自由基,如O2+eCB-→O2·-,O2·-+H++e-→H2O2,H2O2+Ti-OH→Ti-O·+OH·+H2O[18]。在一些催化体系中,如添加HCl,还存在Cl·引发环己烷分子产生环己基自由基的途径[30,34,56]。关于选择性氧化产物,一般认为经环己基过氧化氢C6H11OOH 中间产物分解产生环己酮或环己醇和环己酮,即C6H11OOH→C6H10=O+H2O 和C6H11OOH→C6H10=O+C6H11OH+H2O。环己基过氧化氢一般经下列反应生成:C6H11·+O2→C6H11OO·;C6H11OO·+C6H12→C6H11OOH+C6H11·。也有文献认为环己基过氧化氢可不经环己基自由基而生成,如图6 所示,在V2O5-TiO2光催化体系中,在水分子经空穴氧化产生OH-和H+物种,以及电子经V5+→V4+→O2传 递 生 成O2·-的 共 同 作 用 下,OOH·和环己烷分子生成环己基过氧化氢中间体,按两种途径分解得到环己酮和环己醇产物[16]。关于活性自由基再生,一般认为使环己烷分子活化的活性自由基为OH·,其再生过程主要通过O2的还原反应,如Carneiro 等[11]指出,TiO2晶格氧,H2O 和O2在光辐射下,再生产生OH·([O]晶格H2O+O2→OH·(ads))。OH·再生反应中的H2O可来自于前面光催化反应中的生成水,或催化剂表面的吸附水。催化剂失活是由于催化剂表面被过度氧化产物如碳酸盐或羧酸盐覆盖,由于环己酮/醇没有及时从催化剂表面脱附,继续氧化、开环、脱水生成[11]。
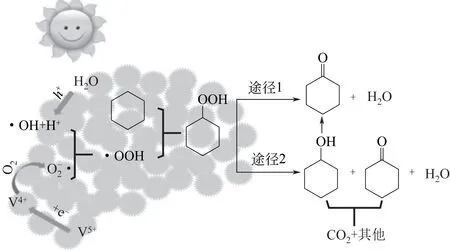
图6 V2O5-TiO2催化体系中光催化环己烷选择性氧化机理[16]
综上所述,反应机理研究中较一致的观点如下:①环己基过氧化氢是中间产物,分解可获得环己酮或环己酮和环己醇;②选择性氧化的反应活性氧自由基为OH·;③未及时脱附的环己酮或环己醇的开环脱氢进一步氧化产生的碳酸盐或羧酸盐沉积于催化剂表面,会导致催化剂失活。尚有分歧或未完全阐明的地方主要有:①单加氧产物中的O 是来源于O2还是催化剂晶格[O]或表面吸附H2O?②产物中生成CO2的活性氧自由基是否为O2·-或其他活性氧物种?
2 光催化环己烷选择性氧化反应的影响参数
影响光催化环己烷选择性氧化的反应参数主要有入射光波长和光强、氧化剂、反应溶剂和水分等。
2.1 反应媒介和光辐射条件的影响
Boarini等[55]报道了TiO2光催化环己烷选择性氧化中溶剂和氧的影响。表2表明,光催化环己烷选择性氧化反应具有溶剂依赖性,添加极性溶剂CH2Cl2会显著增加选择性氧化产物(醇+酮)的产率,抑制CO2的生成,同时随环己烷/CH2Cl2中CH2Cl2含量的增加,环己醇生成速率高于环己酮速率,导致产物中醇/酮比增加[55]。产物分析表明除CO2外,混合溶剂中主要副产物是联环己烷(源于环己基自由基的耦合反应),说明环己基自由基含量相对较高,证明CH2Cl2的加入增加了环己烷的反应活性。另外,从吸附数据来看,溶剂的性质会改变反应物和中间体的表面吸脱附平衡,CH2Cl2加入导致环己醇表面吸附减少,更多保留在反应液中,而环己酮的吸附不受影响,因而导致醇/酮比增加[55]。
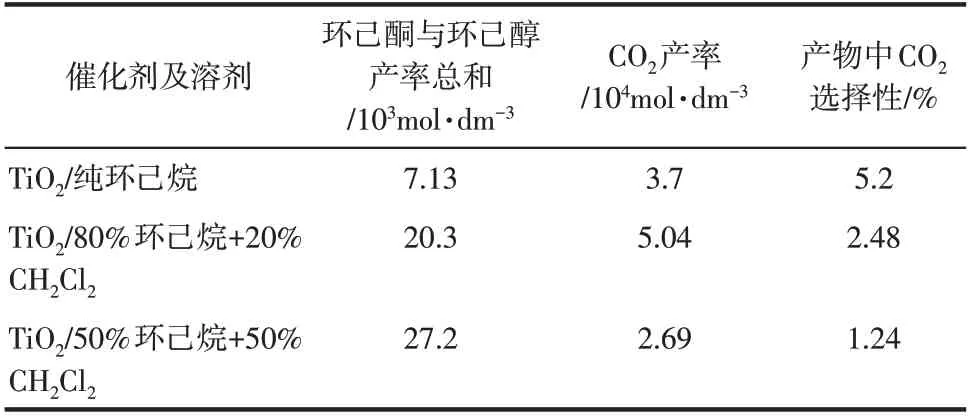
表2 TiO2光催化环己烷选择性氧化中溶剂的影响[55]
Fu等[57]报道了可见光下金属氯化物对环己烷选择性氧化的催化作用,催化剂必须在乙腈或丙酮为溶剂条件下才产生催化活性,主要生成环己醇、环己酮、氯代环己烷、环己烯等产物,金属氯化物和溶剂的弱配位作用起关键作用,添加浓HCl显著地促进催化活性和氯代环己烷的选择性。
Mul 等[14]讨论了入射光波长和TiO2结构对TiO2光催化环己烷选择性氧化反应的影响,指出当无催化剂存在,并以小于270nm波长光源辐射时,环己烷主要为自由基机理的光解反应,产物主要为环己醇,并在反应器壁上产生低聚碳沉积物;加入TiO2并采用Pyrex 材质反应器截除小于270nm 波长光,反应转变为光催化表面反应,选择性氧化产物环己酮主要通过光催化途径生成,因此环己酮选择性高于环己醇选择性;当TiO2晶体结构改变时伴随光吸收波长改变和表面Ti—OH 数量改变,从而影响其催化活性。Grela 等[58]则证实在TiO2光催化环己烷选择性氧化反应中,催化活性受光波长和强度的共同影响,在光波长范围(254~366nm)和光通量范围[0.3~5neinstein/(cm2·s)],产物选择性和产率是和两者共同关联的函数,光波长和光通量的改变会引起反应历程的改变。
2.2 反应气氛的影响
除了在空气/O2环境下进行的光催化环己烷选择性氧化反应,也有部分文献探索了其他反应气氛对光催化环己烷选择性氧化的影响。Ide等[15]发现,在反应体系中加入CO2对环己酮产率和选择性有一定影响,他们以FeO@TiO2为光催化剂,当加入CO2分压为51kPa时,在λ>320nm模拟太阳光辐射24h条件下,环己酮和环己醇总TON(以催化剂中铁含量为计算基准)可从无CO2添加的5.3 增加至206。但如果截除小于420nm 波长的光后,添加CO2并不能引起环己酮和环己醇总TON 明显增长(体系中含51kPa 的CO2时,TON 值从5.3 变化到6.3),说明添加CO2在紫外光下有明显的促进作用。CO2的促进作用在于CO2可吸附在氧化物表面,促进表面吸附的选择性氧化产物环己酮和环己醇脱附,避免进一步反应,或者溶入液相反应体系的CO2可改变反应液的极性,从而影响反应产物的吸附行为[15]。
在另一篇文献中,Fu等[59]以1atm的N2O为氧化剂,钒取代磷钼酸为催化剂,HCl为助催化剂,少量水添加的反应条件下进行可见光激发的光催化环己烷选择性氧化反应,在最优化条件下,环己烷转化率为20.9%,环己酮选择性70.7%,环己醇选择性21.6%,副产物氯代环己烷选择性7.7%。而同一课题组也以相同催化剂在O2、空气和N2条件下进行光催化环己烷选择性氧化反应,发现纯氧环境下,环己烷转化率最高(32.2%)[30]。总之,氧化反应气氛的催化剂反应性能比惰性气氛更高,CO2的添加与否对环己烷选择性氧化有促进作用还需进一步探索。
2.3 水分的影响
Carneiro等[60]对比了在干燥空气和饱和水蒸气的湿空气条件下,TiO2催化的光催化烷烃选择性氧化反应。在干空气环境下,TiO2更易表面积聚碳酸盐和羧酸盐等副产物,减少了活性表面·OH 中心数量,容易迅速失活;湿空气或氧气环境可以促进环己酮在表面的脱附,促进碳酸盐和羧酸盐的分解,从而显著地降低TiO2的失活速率;但水分的影响和TiO2的表面性质有关,具有高结晶结构的TiO2晶粒,适量水分能有效避免团聚,促进表面催化循环,使表面碳酸盐或羧酸盐等副产物转变成CO2而产生“表面自洁”功能,减缓TiO2失活;而对于比表面积较大或缺陷较多的非结晶TiO2光催化剂样品,添加少量水分会在TiO2表面强吸附,导致TiO2晶粒团聚,反而会使催化活性下降[60]。在另一催化体系XPMo12-nVnO40-HCl(X=H+或季铵阳离子)中,水分会干扰质子化活性物种PA[POM-(VVOHM+)Cl-]的形成,因为活性物种PA是由杂多酸盐的VV-O-M中心和HCl经配位作用形成的,水分子会和HCl竞争配位点,影响活性物种形成[30]。
Dijk等[61]采用内置光源的整体光反应器研究了水蒸气对环己烷光催化氧化反应的影响,3μm TiO2涂覆于孔性骨架材料内壁作为整体催化剂,内置光源以光纤均匀分布穿过骨架材料。在含10%~20%O2/N2混合干气环境下,产物中环己酮选择性(>90%)远高于环己醇,催化剂约在80min内失活,在空气下450℃处理可使整体催化剂再生,在相对湿度>20%条件下,连续反应7h 没有观察到催化剂失活,催化剂吸附研究表明反应气中适量水蒸气能促进产物在催化剂表面脱附,从而切断了连续氧化反应形成表面失活物种的途径[61]。此外,当湿度从30%增加至90%时,环己酮和环己醇生成速率都随湿度增加而增加,但环己醇和环己酮生成速率比从0.4 增加到1.0,说明水蒸气使环己醇具有更大的脱附速度。但是,反应气中过高的水蒸气的存在也会降低O2的溶解性,因为O2在环己烷中的溶解性是在水中的50倍,当湿度>70%时,和表面被水分子高度占据的TiO2相比,相对干燥表面的TiO2吸收的O2能更快和环己烷直接接触,有利于表面催化反应[61]。
总之,光催化环己烷选择性氧化是多种因素影响的复杂反应过程,其中,光吸收和激发、环己酮/醇的吸附行为是关键因素。
3 动态分析与发展趋势
近十年来,光催化环己烷选择性氧化的研究取得了一定进展。图7为2008—2019年论文数量柱状图。从图中可以看出,近十年来有关光催化环己烷氧化的文章数量整体趋势是逐步增加的,这也说明光催化环己烷氧化受到越来越多的研究团队的重视。目前所报道的催化剂为TiO2半导体、非TiO2半导体、多金属氧酸盐、有机及非金属、表面等离子体复合催化剂等多种催化体系。图8为目前不同催化剂论文数量百分比饼状图。从各种催化剂所占比例来看,其中半导体催化剂所占比例最大,为54.16%;其次为表面等离子体,为25%;占比最小的为多金属氧酸盐以及有机非金属催化剂,分别为10.42%和10.42%。其中绝大部分催化剂催化环己烷氧化选择性达到95%以上,且存在部分催化剂的转化率达到10%以上。从现有文献来看,光催化环己烷氧化的研究目前还处于实验室研究阶段,达到工业应用的标准还远远不够;其次,未来的研究重点应该为TiO2催化剂的改性以及其他半导体催化剂,同时还应该继续拓宽催化剂的种类,以期找到对光催化环己烷氧化反应效率更高的催化剂。
4 结语

图7 2008—2019年光催化环己烷氧化论文数量

图8 光催化环己烷氧化研究中不同催化剂占比
光催化环己烷选择性氧化生成环己酮/醇能有效利用太阳能,在常温常压温和条件下使难反应的环己烷分子活化,生成具有重要应用价值的单氧化产物酮或醇,和传统热氧化路线比较,在节约化石能源和环保上有突出的优势。近10 年来光催化环己烷选择性氧化的研究取得了一定进展,发展了包括金属氧化物半导体、杂多酸盐、非金属类半导体、表面等离子金属复合催化剂等多种催化体系,某些催化剂在可见光吸收区催化环己烷转化率可达10%,环己酮/醇总选择性可达95%以上,已经优于环己烷热氧化反应催化剂。但光催化环己烷选择性氧化目前尚停留在实验室阶段,尚需在提高催化剂光化学稳定性、光催化剂量子效率、工业规模化光催化反应装置上取得突破。光催化反应机理研究在活性氧自由基、关键中间产物、单加氧产物生成途径上有一致的观点,但关于O2的作用、产物中O进入的途径、深度氧化生成CO2的活性物种等方面尚存在分歧。今后的工作重心仍是大力开发全太阳能光谱响应高效光催化剂,在现有工业化环己烷氧化反应装置基础上改造工业光催化合成装置。
