计算机模拟技术在生物质转化中的应用研究进展
2020-08-17郭鹏坤李攀常春徐桂转石晓华白净方书起
郭鹏坤,李攀,2,常春,3,徐桂转,石晓华,白净,2,方书起,2
(1 郑州大学化工与能源学院,河南郑州450001;2 河南省杰出外籍科学家工作室,河南郑州450001;3 浙江大学生物质化工教育部重点实验室,浙江杭州310027;4 河南农业大学机电工程学院,河南郑州450002)
生物质可提供绿色、清洁的可再生能源。近年来,清洁生产和可持续发展面临的压力越来越大,生物质作为替代能源的重要性日益凸显[1]。生物质“变废为宝”的概念引起了学术界和产业界的广泛关注。通过不同转化方法可以将生物质转化为生物燃料,如生物乙醇、生物柴油、丁醇等;生物质基平台化合物,如丁二酸、谷氨酸、乙酰丙酸、甘油、木糖醇等高附加值产品,已成为当前研究的热点课题。目前,关于生物质转化的研究缺少在大量试验研究的基础上结合试验数据进一步的理论计算,通过对反应机理的深入研究选择生物质转化最优的条件。为此,研究者们尝试结合计算机模拟技术,如流程模拟、分子模拟、条件优化模拟等来强化对生物质转化的研究。与传统试验研究手段相比,计算机模拟是在计算机上实现虚拟的反应过程,并不涉及实际装置的变动,因此使其可以自由地在计算机上进行不同方案、工艺的分析,不仅可以节省时间,也可以节省操作费用;同时计算机模拟还可对经济效益、过程优化、环境评价进行全面的分析和精确评估;并可对复杂过程的规划、研发与开发及技术可靠性做出分析。此外,近年来随着量子化学在理论计算中的广泛应用,研究者们可以从分子水平深入理解生物质转化的本质,研究反应的每一个基本步骤,进而找到反应的路径,找出影响反应速率的关键点。因此计算机模拟技术在生物质转化领域展现出了广阔的应用前景。本文针对计算机模拟技术在生物质转化中的应用,综述了近年来计算机模拟技术在生物质转化中的应用研究进展,为计算机模拟技术在该领域的应用提供参考借鉴。
1 生物质转化类型
生物质转化技术主要有物理转化技术、生物转化技术、热化学转化技术。如图1所示,最常见的物理转化技术是通过压力作用将松散、低热值的生物质转化压缩成型获得固态燃料;生物转化技术主要是利用酶、细菌或微生物将生物质转化为生物燃料,例如,利用厌氧发酵和生物酶分别产生沼气和乙醇;热化学转化技术主要是通过加热和化学相互作用,将生物质转化为燃料或其他高附加值化学品[2]。通过这些转化方式,可以将生物质转化为固体、液体或气体等形式,用于固体燃料、气体燃料和液体燃料,生产电力、化学品等[3],并对相关常用方式作简单介绍。
1.1 生物质气化
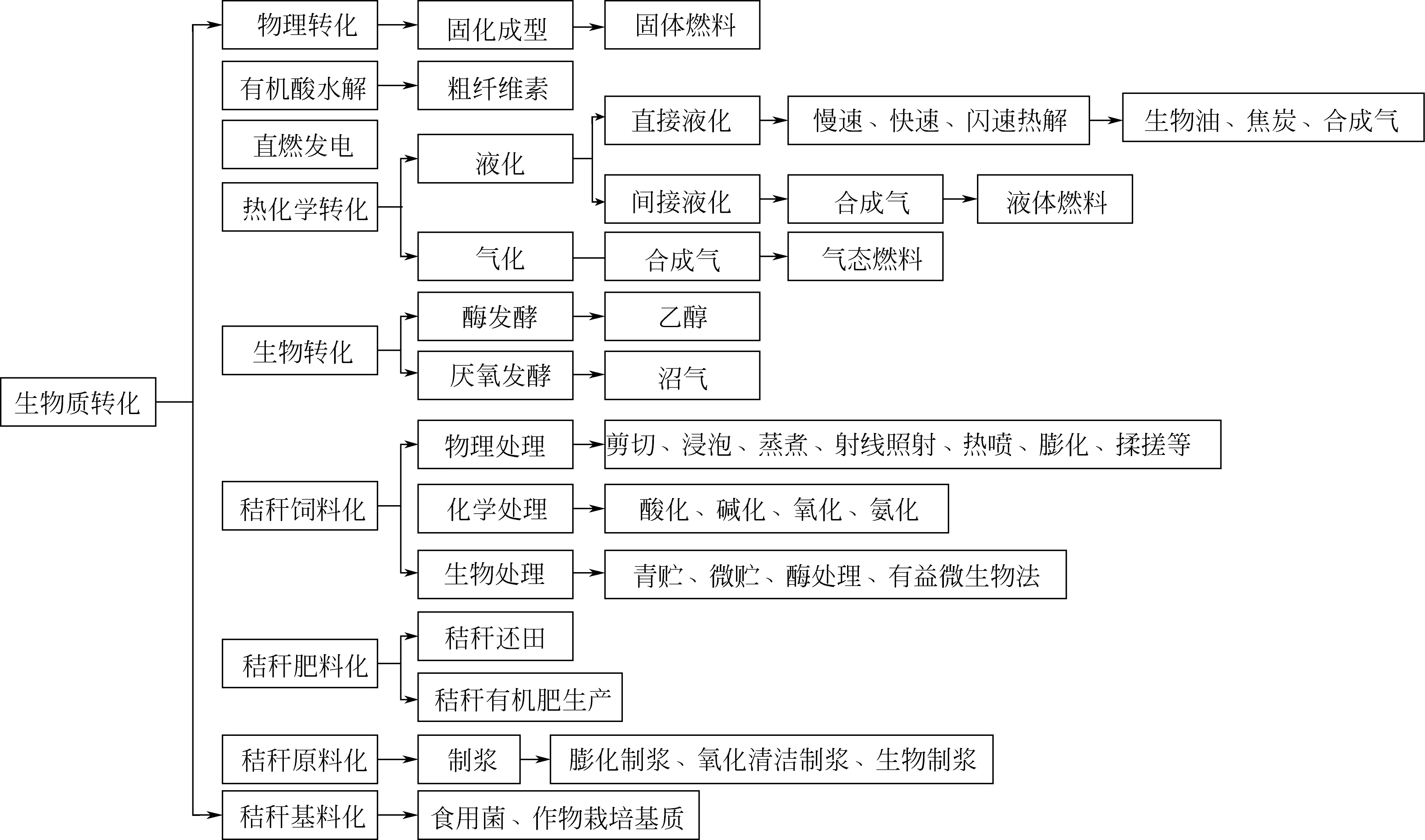
图1 生物质主要转化利用技术
1.2 生物质液化
液化是将固体生物质的大分子结构分解成分子片段,以生成液体产物作为目标[9]。液化过程涉及多种分解反应,如解聚、再聚合、醚键裂解、脱甲氧基化、氧化、异构化、脱水、水解[10]。根据液化方式的不同可分为直接液化、间接液化[11]。其中热解是最早研究的直接液化方法,根据原料在反应器内的升温速率和停留时间,热解可分为慢速热解(升温速率<10℃/s、停留时间>10s)、快速热解(升温速率>10~200℃/s、停留时间=0.5~10s)和闪速热解(升温速率=103~104℃/s、停留时间<0.5s)3种类型。这些产品的组分取决于多种因素,如生物质类型、热解温度、升温速率、停留时间等[12-13]。受升温速率的影响,慢速热解产物主要是固体半焦状,快速热解由于反应速度快、生物油收率相对较高,是目前的首选途径[14]。除此之外,影响生物质热解的因素还有很多,如纤维素的聚合度和晶体形态、半纤维素的多糖和侧枝,以及木质素基本单元中的醚键等都是影响生物质热解反应动力学、中间体演变、最终产物分布的主要因素[15]。研究发现,在温度略温和(350~500℃)、停留时间较短的情况下,有利于生物质转化为液体产品,而较短的停留时间和较低的温度则使生物质主要转化为木炭。表1总结了干木材慢速热解和快速热解的产品分布[16]。
在液化初期,纤维素、半纤维素、木质素的大分子被分解成胶束状的碎片,然后通过一系列反应将这些碎片分解成较小的物质,例如脱水(除去H2O)、脱氢(除去H2)、脱氧(除去O2)、脱羧(除去CO2)以及脱氨(除去氨基酸)。这些化合物一旦形成,可以通过缩合、环化和聚合重新排列形成新的化合物。通过改变操作条件(温度、停留时间)、添加催化剂和溶剂会改变上述反应路径,从而改变合成液体产品的组成[17-18]。Ma等[19]研究了使用木质纤维素生物质作为可持续生产平台分子原料的定向液化,结果表明通过定向液化和竹材生物质的高效分馏,可以同时获得单糖和芳香产物,其中单糖收率为40%,芳香产物收率为20%。

表1 干木材慢速热解和快速热解的产品分布(质量分数)
1.3 生物质物理转化
物理转化技术是通过压力作用将松散、低热值的生物质转化压缩成型获得高能量密度的固态燃料,其步骤主要包括机械铣削、挤压和辐射[20]。通过机械粉碎可以降低生物质颗粒尺寸,对于经过挤压成型的生物质而言,极大地缩减了原有体积,使其更加易于运输和保存,而且所得产品可直接作为固态燃料使用,其品质与中等煤炭相当,但与普通煤炭相比燃烧产生的烟雾更少;微波处理可以去除生物质中的木质素与半纤维素,对进一步的水解(特别是在碱液中)十分有利[21]。
1.4 生物质生物转化
生物转化技术是利用酶、细菌或微生物将生物质转化为生物燃料,主要包括厌氧发酵和生物酶催化。厌氧发酵是利用微生物代谢活动将生物质分解为沼气,用于燃烧、发电等。因其投资较大、产量低,大多应用于污水处理厂和有机易腐物垃圾堆肥的处理过程。而以醋渣为酸化剂,对玉米秸秆进行预处理是一种高效厌氧共发酵的新方法。研究发现,在150℃条件下,经过预处理共发酵的甲烷产量比未预处理的甲烷产量高35.7%左右[22]。然而,Cheng 等[23]在青贮草发酵生产氢气和甲烷的研究中发现,经过酸预处理的青贮草氢气产率与处理的氢气产率相比增加了3倍,但在第二阶段的发酵过程中,未经处理的青贮草的甲烷产量较低,导致总能量转化率下降。生物酶催化主要是在纤维素酶的作用下,将纤维素、半纤维素转化为乙醇,生物酶催化过程主要包括:①原料的预处理;②生产纤维素酶;③纤维素水解糖化;④半纤维素、纤维素水解产物戊糖和己糖的发酵。由于生物质中木质素会阻碍纤维素酶与底物的接触,导致生物质酶催化转化速率较慢,且目前缺少较为经济可行的戊糖发酵技术,这些都是限制酶催化纤维素类生物质生产燃料乙醇等的难题[24]。
1.5 有机酸水解技术
有机酸水解技术是以甲酸、乙酸等有机酸作为混合溶剂处理木质纤维素,以获得粗纤维素产品为目的的生物质转化技术[25]。Jeroen 等[26]使用甲酸、乙酸的混合溶剂处理小麦秸秆得到了粗纤维素产品,其中大多数木聚糖(67%)和木质素(96%)被溶解,生成的纤维素纸浆有93%是最初的纤维素,纯度为63%。经过有机酸处理后得到的粗纤维素产品可进一步通过酶水解等技术转化为生物乙醇、生物乙烯。
2 流程模拟在生物质转化中的应用
自20世纪50年代起,人们开始采用计算机模拟技术解决化工过程中的数学问题,目前流程模拟技术也成为研究者们普遍采用的研究方法。流程模拟实际上是利用计算机程序定量计算一个化学过程中的特性方程。其主要过程是采用适当的模拟软件,将由多个单元操作组成的化工流程用数学模型描述,模拟实际的生产过程,并在计算机上通过改变各种有效条件得到所需要的结果。模拟涉及的数据一般包括进料的温度、压力、流量、组成、有关的工艺操作条件、产品规格以及相关的设备参数。其中Aspen Plus(advanced system for process engineering)在生物质转化中颇具应用价值。
2.1 Aspen Plus在生物质转化中的应用
Aspen Plus 由美国Aspen Tech 公司研发,基于稳态化工模拟、优化、灵敏度分析和经济评价的大型化工流程模拟软件。为用户提供了一套完整的单元操作模块,可用于各种操作过程的模拟以及从单个操作单元到整个工艺流程的模拟。Aspen Plus 是唯一能处理带有固体、电解质、生物质和常规物料等复杂体系的流程模拟系统。由于生物质在转化过程中涉及的反应较多且较为复杂,使得模拟计算的难度大大增加,因此建立可靠性更好的模型成为研究人员的目标。将Aspen Plus在生物质热转化中转化方式、反应器类型、Aspen plus 模型的不同进行比较,如表2所示。
Aspen Plus 模型主要包括热力学模型和动力学模型。热力学模型是基于生物质转化过程中的质量守恒和能量守恒,假定在合成气离开反应器之前,气化过程中的所有反应都达到化学平衡状态,在吉布斯自由能最小的情况下计算系统混合气的组成(也叫minimizing gibbs free energy model,MGFE 模型),但这一模型将气化炉简化成一个反应器,忽略了气化炉的结构、气体流动状态以及其他动力学相关参数,因此该模型通常需要设置相关限制条件来修正模型。闫桂焕等[27]以木屑为原料,将系统散热和碳的不完全转化等因素纳入到热力学模型中,研究了气化剂温度、空气当量比对气化结果的影响。Jarungthammachote 等[28]建立了单室气化炉热力学模型,对产气组分进行了预测后发现模拟结果中CO 和CO2与实验数据偏差较大,考虑到碳转化的影响,于是对温度进行了修正,通过约束方程和能量平衡计算来修正模型,进一步缩小了模拟结果和实验数据的偏差。Puig-Gamero 等[29]以热力学为基础设计了一种双室气化炉,它可以使气化区和燃烧区分离,使焦炭在燃烧区燃烧,为气化区供热,不仅提高了能量的利用,而且获得了高H2、CO 占比的合成气。此外,为了提高生物质气化工艺的经济可行性,一些研究者将气化与生物发酵工艺结合,该模型由生物质气化、发酵、产品回收3个模块组成[32]。另外有研究对合成气进一步净化,在一定条件下与CO、H2O作用生成甲醇、乙醇等[33]。

表2 Aspen Plus在生物质热转化中的应用
动力学模型是将反应流体力学特性和动力学特性纳入到模型中,而且模型中的动力学速率表达式是由实验数据拟合得到,虽然该模型计算量较大,但模拟可靠性大大增强。Yu 等[30]建立了一种基于生物质综合气化动力学的反应模型(RXN 模型),该模型将炭的气化分为多相反应和均相反应,分别设置气固两相反应装置的停留时间,并将不同当量比下的合成气组分预测值与相关实验数据进行了比较,验证了动力学模型的正确性。Sadhwani等[31]尝试以CO2作气化剂,用一个动态链接的FORTRAN子程序包含了Boudouard 反应的动力学模型。该模型对同一气化过程的多个阶段分别建模,然后组成一个单独的模拟过程。然而该模型对Boudouard 反应碳转化的预测远低于实验数据。文献中也报道了类似的关于热力学平衡模型达到更高碳转换水平的研究结果[34-35]。为了进一步提高动力学模型在碳转化过程的可靠性,Kaushal 等[36]建立了焦油生成和裂解的子模型,将结果与实验数据和其他仿真结果进行了比较。结果表明,定义焦油及其动力学显著提高了模拟结果的可靠性。
Aspen Plus 在生物质热解过程也有应用。如Peters 等[37]提出了一种新的生物质热解动力学反应模型,该模型使用149个单独的反应来模拟生物质的挥发、分解和重组过程。生物油采用高水平的细节建模,使用多达33 种模型化合物,可以全面估算生物油的性质并预测进一步的升级反应。如图2所示,通过Aspen Plus再现实验热解过程,得到的馏分产率(生物油、炭和气体)、含水率和热解产物的元素组成具有很高的一致性。此外,Wright等[39]使用Aspen Plus 快速热解模型来模拟过程的质量和能量平衡。该工艺使用玉米秸秆作为原料,包括了从生物油蒸馏到石脑油(汽油混合物)和柴油馏分的步骤。为了进一步提高模型的可靠性,Humbird 等[40]将稳态热解反应器模型与Aspen Plus中更大的稳态过程模型相结合,实现了一种具有详细反应动力学和一维流体动力学的生物质快速热解反应器模型,这种将详细的反应器模型与稳态流模型相结合的方法,也有利于开发更加准确的技术经济分析。另外,近年来也有通过分别定义热解过程中的气相、液相组成来提高模拟结果可靠性的报道[38]。
Aspen Plus 在生物质生物转化过程也有应用,如生物质发酵制备纤维乙醇。该方法目前已得到工业化应用,不少研究者通过建立生物质生物炼制全过程流程模拟模型,来实现对产业化纤维乙醇生产技术的技术经济评价、工艺优化。如乔庆安等[41]在构建了该过程相关物质物性数据库的基础上,建立了年加工30 万吨玉米秸秆生产纤维乙醇的热力学模型,该模型考虑了水用量、蒸汽能耗等工程问题,可以对相关工艺设计和优化提供参考。Kristin等[38]将Aspen Plus 的生物精炼厂不同原料和转化途径的工艺模型与整数线性规划(mixed-integer linear programming,MILP)模型相关联,用于详细的技术经济和环境性能分析,结果表明,改进转化技术是克服纤维素乙醇商业化障碍和纤维素生物燃料扩大社会影响的关键问题。Aspen Plus 在综合利用系统方面也有应用。Damartzis等[42]对由流化床气化反应器、气体净化系统和发电用内燃机组成的小型热电联产生物质气化系统进行了研究。Sadhukhan 等[43]建立了基于过程模拟和集成的生物质气化燃料电池(BGFC)系统集成设计方法,并与生物质气化联合循环系统进行了能量比较,结果显示综合BGFC 系统的发电潜力是综合BGCC 系统的两倍。综合系统的应用不仅提高了能量的利用率,而且与单一系统相比具有更好的经济性。

图2 Aspen Plus模拟生物质热解的工艺流程[37]
2.2 PRO/Ⅱ在生物质转化中的应用
PRO/Ⅱ是另一款被广泛使用的流程模拟软件,该软件由美国科学模拟公司(SimSc)研发推出,软件主要由化学组分库、热力学方法库、单元操作模块、热力学数据管理器(TDM)和伪FORTRAN编程器组成。与Aspen Plus相似,该软件的模拟计算方法也是采用序贯模块法,即通过给定的物流数据和单元设备参数计算输出物流的相关参数。与其他化工模拟软件相比,PRO/Ⅱ由于其发展历程较长,因而积累了大量工程上的实际经验数据,也使得其模拟结果能较好地符合工程上的实际结果。
PRO/Ⅱ也可以模拟生物质气化的复杂过程。通过分析生物质复杂的组分,可以确定生物质的化学分子式和可燃部分热值,在定义流程模块的基础上,可进一步定义气化的流程图[44]。郝巧玲[45]分析了稻草、玉米秸秆、高粱秸秆、松木等生物质的组分以及单位质量的发热量,建立了生物质与煤共气化的反应流程模型。探究了温度、压力、煤与生物质配比等因素对共气化特性的影响规律。在生物质热解方面,PRO/Ⅱ也有应用,为了评价由桉树热解与发电厂组成的热电联产系统的经济可行性,Anna等[46]在PRO/Ⅱ软件中开发并模拟了每天2000t桉树热解和发电厂的过程模型,如图3所示。该过程模型由生物质培养与预处理(U1)、生物质快速热解(U2)和生物油发电(U3)组成,该模型以质量和能量平衡为基础,并以此作为技术经济分析的基础,模拟结果表明,该技术与当前发电成本相比,具有一定的竞争优势。
3 分子模拟在生物质转化中的应用
分子模拟(molecular simulation or modelling and simulation)是20 世纪末兴起的一种计算机模拟技术,随着量子力学的逐步完善、计算机技术的不断发展,近年来分子模拟技术在化学化工、生命科学、材料科学和物理等诸多领域均有应用[47]。因此在实验研究的基础上,有必要进行进一步的理论探索。利用量子化学方法从分子水平上研究反应机理,对于有复杂反应体系特征的生物质转化过程的研究具有极其重大的意义。
3.1 Gaussian 09w在生物质转化中的应用
Gaussian 09w 是当前应用最广泛的量子化学计算软件之一,主要用来计算和推测分子能量和结构、过渡态的能量和结构,计算化学键和反应能量、红外光谱、拉曼光谱、振动分析、核磁谱图、热力学性质、反应路径[48]。其中密度泛函理论(DFT)是量子化学计算方法中性价比最高、应用最广泛的方法。生物质转化的机理相当复杂,特别是在反应中添加催化剂时,这些反应存在着相互影响,将DFT 应用于生物质转化研究中,可对反应过程进行理论分析,有助于从分子水平深入了解化学反应的本质,为解决关键问题提供强有力的理论依据[49]。将Gaussian 09w 在生物质转化中反应物、反应物模型、计算类型、计算方法的不同进行比较,如表3所示。
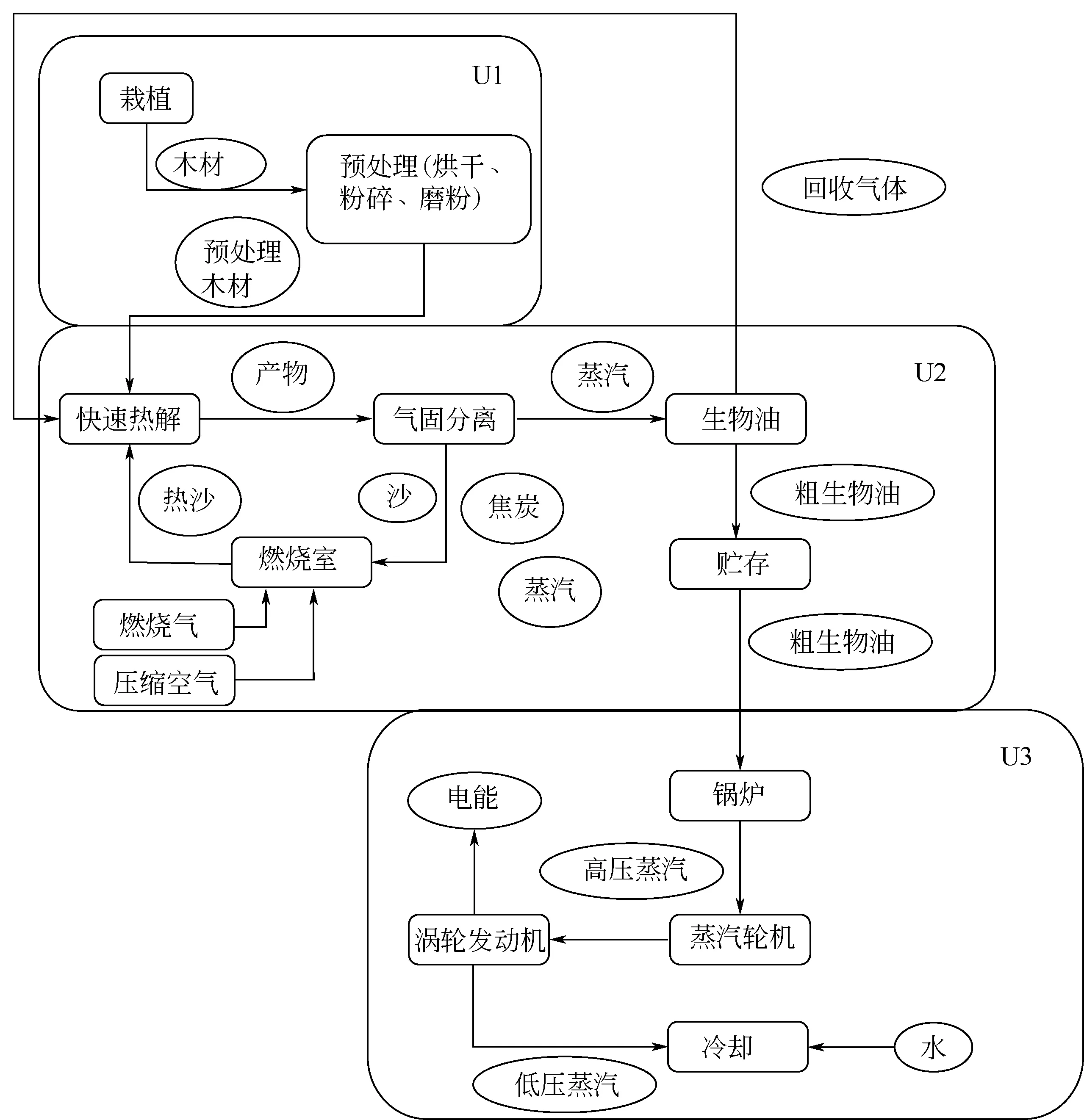
图3 PRO/Ⅱ模拟桉树热解的工艺流程[46]
Gaussian 09w 可构造并优化复杂的生物大分子模型。为了探究纤维素取代基在生物质酸性水解过程中的作用机理,Mu 等[50]以纤维二糖作为纤维素模型,经多次迭代计算取得了纤维素二糖的稳定构型,将取代和未取代的纤维二糖通过M06-2X密度泛函和6-31+G*基组进行DFT 计算。分析了场效应、电荷基团的存在对水解过程的影响。结果表明,水解速率受取代基的场效应影响,这种场效应主要取决于取代基上的负电荷量和带负电荷的基团与氧代羰基之间的距离。Zhang 等[51]采用DFT 研究了以纤维素三糖为模型化合物的纤维素脱水过程,如图4 所示。因为采用纤维三糖作为模型化合物可以考虑相邻葡萄糖单元对吡喃环断裂反应的影响。DFT 研究证实,羟基的位置对纤维素脱水有重要影响。羟基活性最高的是—O2H,其次是—O3H和—O6H。
Gaussian 09w 可以寻找反应过渡态(transition state,TS)。在已知反应物与生成物结构的情况下,对反应物和生成物进行结构优化和频率计算,然后通过本征反应坐标(IRC)验证与正确的反应物和产物相关的过渡态。Zhou等[52]在研究木质素二聚体热分解机理和产物生成途径的过程中,计算并验证了多个过渡态结构。Lu等[53]在探索纤维素单糖在热解过程中羟基乙醛的形成机理时,通过结构优化和频率计算了过渡态,然后采用与优化相同的方法进行频率分析,得到了标准的热力学参数,确保过渡态的正确性。

表3 分子模拟在生物质转化中的部分应用
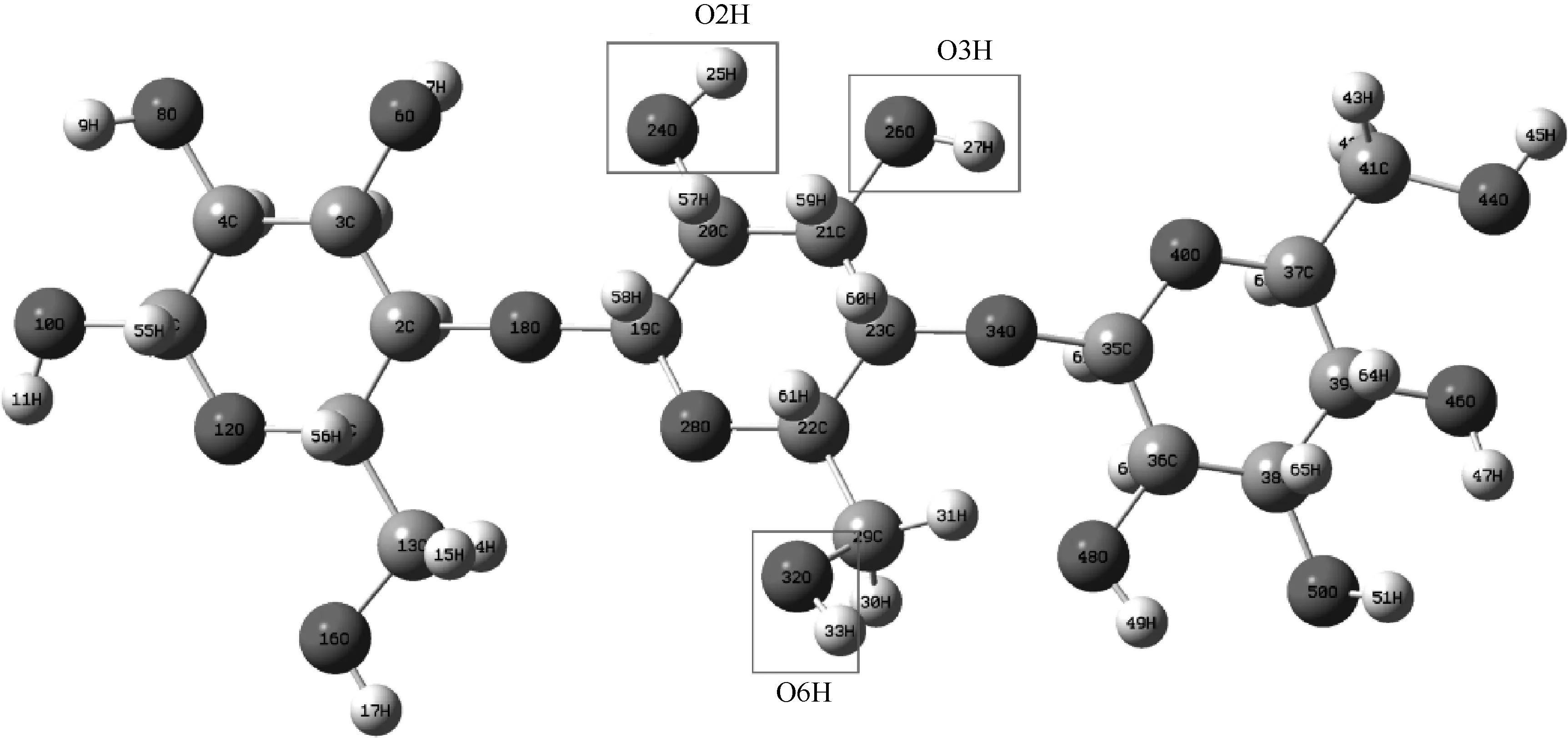
图4 Gaussian构造优化的纤维三糖模型
Gaussian 09w 可以计算热力学参数。在确定了过渡态、中间体结构的基础上,对优化过的反应物、产物的构型进行频率计算,并在频率计算名中加入“temperature”关键词,可计算反应物、产物在不同温度下的热力学参数。黄金保等[54]在进行纤维素热解形成左旋葡聚糖的机理研究时,通过频率计算得到了反应各驻点(反应物、过渡态、中间体和生成物)的总能量(经过零点能校正的能量),并以此计算出反应的焓变和吉布斯自由能变。获得的焓变和吉布斯自由能变可用来判断反应的自发性和产物的百分含量。在生物质转化过程中,多个反应同时进行且互相影响,寻求反应的最优路径成为研究生物质转化机理的难题,而通过计算吉布斯自由能变是目前使用较多的方法。首先,设计由反应物到生成物可能的反应路径,接着将路径中涉及的所有物质(反应物、中间体、过渡态、生成物)进行结构优化,然后计算上述物质的热力学参数,最终获得各物质的焓值和吉布斯自由能。以反应物为基准可计算出反应途径中各反应的吉布斯函数变。如Li等[55]在研究咪唑啉基离子液体催化果糖脱水制备5-羟甲基糠醛的机理时,分别计算并绘制了果糖在3个脱水过程中各物质的吉布斯自由能曲线,如图5所示,在第二个脱水过程中,共同路线(绿线)在反应第二阶段反应活化能垒最低,说明该反应发生的可能性较高,可能导致羟甲基糠醛的生成,相反活化能垒最高(红线)为整个反应的控制步骤,整个催化过程的能垒为41.5kcal/mol(1kcal/mol=4.184kJ/mol),与以往研究(75.6kcal/mol)有明显降低。此外,在多路径反应同时存在时,计算体系的热力学参数对反应路径的研究也有很大意义。如Huang 等[56]在研究β-D-吡喃葡萄糖的热解机理时,提出了4种可能的热解途径,并计算了不同温度下各反应路线的标准热力学和动力学参数,以此绘制了势能剖面图。结果表明,所有反应都是吸热的,当反应温度超过550K 时可以自发进行。反应路径1 和2 中的吉布斯自由能和速率决定步骤活化能的变化小于反应路径3 和4。基于热力学和动力学分析,反应途径1和2是主要的热解反应通道。
3.2 Materials Studio
Materials Studio 是由美国Accelrys 公司研发的商用量化计算软件。包含分子动力学(MD)、量子力学(QM)等多种先进计算方法,主要用于研究和预测材料的相关性质以及解决材料合成和化工研发过程中的问题。Materials Studio 所包含的量子力学模块主要有:ONETEP (线性标度方法)、CASTEP(平面波赝势方法)、QMERA(量子力学/分子力学杂化方法)和DMol3(原子轨道线性组合方法)。与Gaussian 09w 相似的是,DMol3 也是基于密度泛函理论(DFT)的量子力学模块,它通过数值方程来描述系统的电子状态,这种方式能快速模拟材料的化学转化过程以及预测材料的性质。Materials Studio 的使用与Gaussian 09w 也有不少相似之处,如其中的Visualizer 模块、Discover 模块、LST/QST 模块分别对应Gaussian 09w 中分子模型的构造、分子模型的优化和寻找反应的过渡态模块。
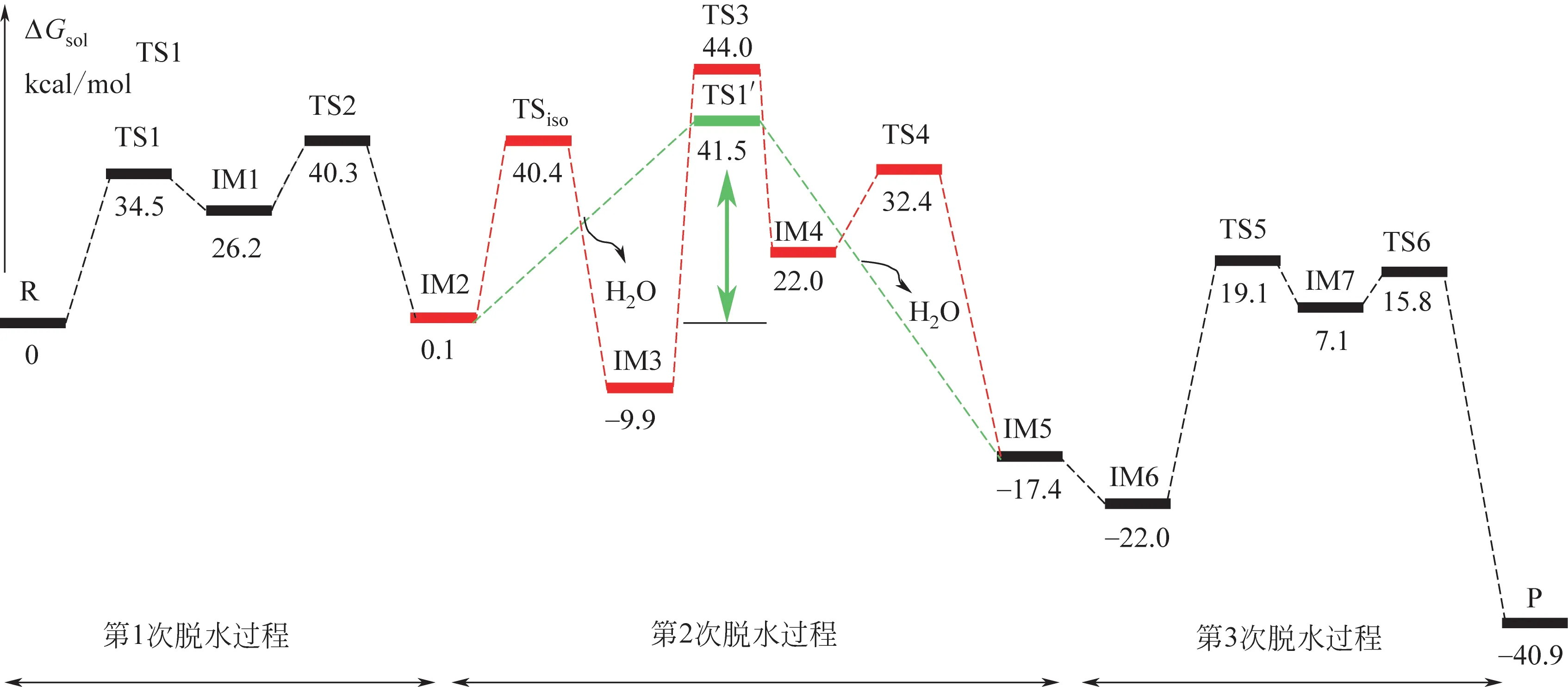
图5 由Gaussian 09w计算得到的果糖脱水过程的吉布斯自由能变化[55]
在生物质转化应用方面,基于DMol3计算模块的研究较多,如梁洪林[57]以左旋葡萄糖作为纤维素模型,对反应过程涉及的多种物质(反应物、中间体、过渡态和生成物)进行了优化计算,通过对23 条可能的反应路径分析后发现,与其他气体相比,纤维素热解更易生成CO。王鹏恒[58]在研究生物质气化过程中混合气在催化剂Pd/Al2O3表面的吸附机理时,采用DMol3 计算模块对Pb 负载于γ-Al2O3表面的吸附进行了系统研究,该研究发现混合气与γ-Al2O3表面吸附作用由强及弱依次为:CO、H2、CH4,为生物质气化过程催化剂的设计和优化提供了一定的参考。
3.3 GROMACS
分子动力学(molecular dynamics,MD)模拟是分子模拟中的重要方法,因此广泛应用于生物、物理、化学、材料、医学等诸多领域[59]。其中,GROMACS是由荷兰格罗宁根大学研发的著名开源分子动力学模拟软件之一,主要用于执行具有复杂成键作用的生物化学分子的模拟计算[60]。由于其代码的实现可以结合硬件特点,使得GROMACS可以对不同的硬件构建进行优化,这种对硬件资源的充分利用大大提升了GROMACS的计算性能。
GROMACS 可进行生物质转化的MD 模拟,主要应用于生物质大分子在溶剂中的分子动力学行为。如Vasudevan 等[61]利用GROMACS 软件分析了葡萄糖在纯水溶剂和3 种混合溶剂[二甲亚砜(DMSO)/水、四氢呋喃(THF)/水和二甲基甲酰胺(DMF)/水]中的分子动力学,研究了溶剂与葡萄糖之间氢键的寿命和活化自由能以及葡萄糖分子的聚集趋势。结果发现,助溶剂和水在葡萄糖周围的这种优先排列可能在促进形成5-羟甲基糠醛(HMF)和乙酰丙酸的反应途径中起作用,并且可以减少葡萄糖降解为不想要的脱水产物的可能性。最近也有关于采用GROMACS进行离子液体降解木质纤维素MD模拟的报道[62-63]。
4 条件优化模拟在生物质转化中的应用
响应面法(response surface method, RSM)是一种数学和统计方法的集合,可以对受多个变量影响的响应进行建模和分析,以得到最优条件的分析方法。与传统试验设计方法相比,RSM 可以有效地利用统计程序调查多个参数的交互作用效果,并通过图形技术显示出这些参数与响应的关系,实现最优条件的可视化[64]。本文作者课题组在先前的工作中曾将响应面优化法应用到生物质转化的研究中来,如Deng 等[65]在利用廉价金属硫酸盐催化小麦秸秆高效转化为乙酰丙酸甲酯的研究中,采用响应面法研究了反应温度、反应时间和催化剂用量对反应收率的影响。三维响应面图如图6所示。由于生物质转化过程中影响因素较多,因此有效地分析这些因素间的交互作用对生物质研究有很大帮助。樊永胜[66]研究了生物质在热解过程中热解温度、体系压力、升温速率和保温时间对生物原油产率的交互影响,结果表明,单一因素对原油产率的影响不是简单的线性关系,热解温度和升温速率对生物原油产率的影响存在显著的交互作用。Tiong 等[67]选择了一种基于中心复合设计(CCD)的响应面法(RSM)来优化乙酰丙酸和乙酰丙酸乙酯产品的工艺条件,研究了在水-乙醇介质中反应温度、反应时间、乙醇与底物体积比的影响及其相互关系,发现在所有交互项中,离子液体与生物量之比和反应温度之间的相互作用在所有相互作用项中最为显著,这显著促进了产生乙酰丙酸的催化反应。

图6 RSM分析的不同因素间的交互影响[65]
5 其他计算机技术在生物质转化中的应用
5.1 人工神经网络
人工神经网络(artificial neural networks,ANNs)是一种模拟人脑神经网络特性,经由许多处理信息的基本单元(神经元)建立的分布式并行信息处理的数学模型。它通过将大量的数据关联起来,并将这些数据不断优化调整来达到处理信息的目的[68]。人工神经网络已成功地应用于天气预报、食品科学、医学、电子工程等领域,然而人工神经网络在生物质热物性和元素组成预测中的应用仍处于发展阶段。
到2000 年为止,大部分模型主要基于线性回归方法,然而生物质的一些近似组分分析与它们的高热值之间的关系是非线性的,因此基于线性回归模型的预测是不够的[69]。不少研究者在对比线性回归和非线性回归两种高热值分析方法时,也得出了基于非线性的模型具有更好的预测效果,如目前广泛使用的多层前馈神经网络MLP(图7),该体系结构包含一个输入层、一个输出层和一个“隐藏”的中间层,其中输入层节点数为自变量个数,输出层节点数为因变量个数,隐藏层的数量和每个隐藏层中的节点数是可调的参数[70-71]。通过对比线性方法与神经网络方法得到的结果,Alex等[72]发现了神经网络预测的生物柴油某些性质的非线性趋势,如生物柴油黏度、碘值和诱导期的预测。为了达到更高的模拟精度,Ismail 等[73]利用人工神经网络(ANNs)建立了替代单元运算模型、热力学模型和混合模型的替代模型,并使用大量的数据来训练神经网络,使得该模型可以准确地捕获下垫模型。结果表明,采用碱催化酯交换反应器可获得最佳工艺流程。这表明使用ANNs作为代理模型来降低模型的复杂性以实现过程合成的目的是很有希望的。Ghugare 等[69]也提出了一种新的人工智能形式来开发生物量高热值预测模型。
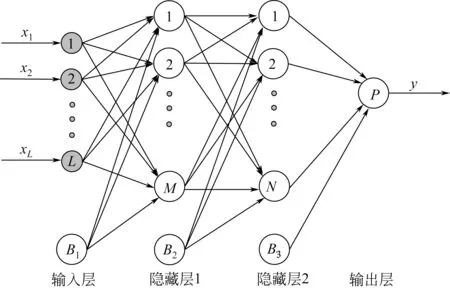
图7 多层前馈神经网络(MLP)结构
5.2 全生命周期评估
全生命周期评估(life cycle assessment, LCA)是用于评价某一产品系统在其整个生命周期内对系统输入和输出的物质或能量及潜在的环境影响的方法。可贯穿产品从原料收集、运输和储存到生产加工、销售,再到回收、废弃物利用与管理的整个过程。从工作流程上划分,生命周期模拟主要包括确定研究目标和范围、清单分析、影响评价和结果解释4个步骤[74]。
除了应用于生物质热解、气化过程中,生命周期模拟还可应用于生物质热电联产系统等综合系统,用于综合分析经济、能源、环境方面的影响[75]。此外,还可用于对同一产品系统不同用途的比较分析,Soam等[76]利用全生命周期分析了秸秆发电、秸秆沼气、秸秆还田和秸秆饲料这4种利用方式,发现秸秆发电和生产沼气对全球升温潜能值、酸化潜能值、光化学氧化剂的创造潜力等的环境效益较高,秸秆饲料对富营养化潜能值的环境效益较高。
5.3 数值模拟
数值模拟是通过求解相关守恒定律和公式来达到深入理解相关规律的方法,主要的模型有热力学平衡模型、化学动力学模型、计算流体动力学模型(computational fluid dynamics, CFD)等,其中CFD方法因其可以运用多种先进数值方法建模,来描述多相反应器中的流体动力学的复杂瞬态行为而受到研究者的广泛使用。如Shi 等[77]采用欧拉-欧拉方法,对用于干燥生物质颗粒的热转化螺杆反应器内的固体流体动力学和固体反混行为进行了三维流体动力学CFD 模拟研究,分析了反应器内固体流动动力学、固体停留时间分布、固体反混程度等对生物质转化过程的影响。此外,还可以使用多种模型相结合的方式来强化模拟结果的可靠性。如Hariswaran 等[78]采用将CFD 方法与化学动力学模型结合的方法,模拟了纤维素的酶解过程。该研究发现纤维素颗粒的沉降和酶的扩散决定了较低混合速率下的总转化率,而在较高的混合速率下反应速率占主导地位。
6 展望
计算机模拟技术已成功应用于诸多研究领域,在生物质转化方面,对于机理的探索和工艺的分析优化都具有重要的意义。针对同一生物质转化过程,不同的模拟方法与欲实现的目的一一对应,每种模拟方法的切入点虽不同,但对于同一生物质转化过程的研究可考虑多种模拟方法的结合,比如在宏观层面工艺的分析和优化,可通过流程模拟Aspen Plus、PRO/Ⅱ等分析系统中物料的温度、压力、流量、组成;通过条件优化模拟Design Expert等分析多个变量与响应的交互关系,以求得最佳的工艺条件;在微观层面对于反应机理的探索,可通过Gaussian 09w、Materials Studio 等构建纤维素、木质素等大分子物质,寻找在实际反应中难以捕捉的过渡态,计算体系中涉及的所有物质(反应物、中间体、过渡态、生成物)的热力学参数,在复杂的反应体系中寻找反应的最优路径;以及通过分子动力学模拟GROMACS等考察生物质大分子在溶剂中的分子动力学行为;亦或在基于大量数据的基础上运用人工神经网络预测产物黏度、热值等信息,通过全生命周期评估分析产品对环境的潜在影响。模拟过程中难免对某一过程简化,而简化意味着与真实结果的偏离,为了进一步提升模拟结果的可靠性,建议在以下方面深入研究。
(1)将生物质反应体系的复杂性纳入到模型中,尽量减少对真实过程的简化,建立更加可靠的模型。
(2)对同一过程的流程模拟,可以采用动力学模型与热力学模型结合的方式来提高模拟结果的可靠性。
(3)分子模拟过程中建立大分子模型时可以选择多种模型分别模拟(如纤维素转化过程中可以选择纤维二糖、纤维三糖作为模型)。
综上所述,计算机模拟技术在生物质转化领域具有重要的应用价值,随着计算机模拟技术的不断发展,模拟结果的可靠性也将逐步提升,模拟技术在生物质转化中的应用将成为重要的研究手段。
