幼年巨大型黄色肉芽肿1例
2020-08-17林杨杨左海亮刘欣欣李钦峰
林杨杨,左海亮,赵 丽,刘欣欣,秦 蓓,李钦峰
幼年黄色肉芽肿(juvenile xanthogranuloma,JXG)又称痣样黄色内皮瘤(nevoxanthoendothelioma),是一种具有自限性的良性组织细胞肿瘤,由Adamson[1]于1905年首次报告,主要发生于婴幼儿及儿童,临床非常罕见。笔者对我院皮肤科收治的1例JXG 患儿的临床特征及病理表现进行总结,旨在对临床医师避免漏诊及误诊提供借鉴。
1 病例报告
患儿,女,4个月,因左上肢黄红色肿物2个月,于2018年2月至天津市儿童医院皮肤科就诊。家属主诉,患儿出生后2个月左上肢无明显诱因出现一个黄豆大淡红色结节,近期肿块逐渐增大,无红肿热痛(图1A)。发病后患儿精神、食欲、睡眠及大小便均正常,父母非近亲,家族无类似病史。左上肢见直径2.5 cm的黄红色半球形肿块,突于皮面,质地坚韧,边界清楚,结节中央可见暗褐色的结痂;Darier征阴性,黏膜未见异常。体格检查:发育正常,营养良好,全身浅表淋巴结未触及肿大,系统检查未见异常;眼科检查未见明显异常。实验室检查:血、尿常规、肝肾功能、血糖、血脂均未见明显异常。胸部X线片及腹部超声未见明显异常。组织活检:局部麻醉下钻取患儿部分皮损组织用4%甲醛溶液固定并用石蜡包埋后,将其切为5 μm的切片并行苏木精和伊红(HE)染色,脱蜡至水后,进行免疫组化染色(图1B、C),HE染色示表皮大致正常,真皮全层见致密的组织细胞浸润,夹杂嗜酸性粒细胞浸润,部分区域组织细胞胞质淡而空,核较小,呈圆形或短梭形,未见Touton细胞、多核巨细胞,免疫组化提示CD68(+)、CD163(+)、cycinD1(+)、CD1a(-)、S-100(-);特殊染色:PAS染色未见真菌菌丝。病理诊断:考虑JXG。因本病具有自限性,未予特殊治疗,建议出院后随访。6个月后,家属要求再次进行病理活检,术后病理示:真皮内可见大量组织细胞弥漫性浸润,其间可见大量泡沫状组织细胞浸润,并有Touton巨细胞散在分布(图1D)。结合病灶大小及病理出现典型的Touton巨细胞,最终诊断:巨大型JXG。继续随诊观察,随访至2020-02,皮损渐消退,其他系统检查未见明显异常。
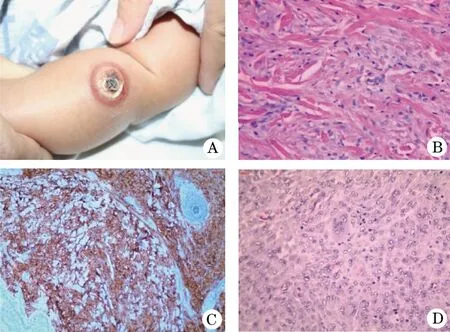
图1 幼年巨大黄色肉芽肿患者皮损表现及病理特点A.左上肢直径2.5 cm的黄红色半球形肿块,突于皮面,质地坚韧,边界清楚,结节中央可见暗褐色的结痂; B.真皮全层见致密的组织细胞浸润,夹杂嗜酸性粒细胞浸润(HE,×400);C.CD68(+)免疫组化(×400);D.真皮内可见大量组织细胞弥漫性浸润,其间可见大量泡沫状组织细胞浸润,并有Touton巨细胞散在分布(HE,×400)
2 讨 论
JXG属于非朗格汉斯细胞组织细胞增生的一种[2],是一种极为罕见良性疾病,病因尚不明确,本病相关文献仅为个案报道。90%的JXG患者为儿童,男女比例为1.5∶1,成年患者无性别差异。皮损常发生于6个月以内婴儿,但有10%~20%出生时即可发现,好发于头面部、躯干及四肢,为隆起性黄色皮肤丘疹或结节,数量1个至数个不等,边界清楚。单发结节较大,多发结节较小,质地较柔软[3]。本例病灶较大,直径达2.5 cm,文献[4]报道非常少见。结节一般在患儿2~6岁时自然消退,可遗留少许色素沉着,但成人常较持久。本病绝大多数单独发生于皮肤[5],但也可累及身体其他器官,如眼、中枢神经系统、肝脏、肾脏及口腔黏膜等[6-8],累及皮肤以外组织和器官称为系统性JXG[9],儿童JXG最常见受累的部位是眼睛[10],可波及睫状体严重者导致失明,本例随访2年未见除皮肤外的其他系统受累。
本病临床鉴别诊断主要包括朗格汉斯细胞组织细胞增多症(langerhans’cell histiocytosis,LCH)、良性头部组织细胞增多症(benign cephalic histiocytosis,BCH)、肥大细胞瘤(mastocytoma)、泛发性发疹性组织细胞瘤(generalized eruptive histiocytoma,GEH)及结节性黄瘤(xanthomatuberosum,XT)等。肥大细胞瘤在摩擦后出现风团,组织病理中可见肥大细胞。而多核巨细胞和泡沫细胞可以鉴别JXG和BCH、GEH。LCH免疫组化S-100蛋白和CDla均阳性,且可以通过电镜检测到Birbeek颗粒。XT常有脂质代谢紊乱,好发于关节及四肢伸侧,皮损较少自行消退。
由于JXG发病率非常低,临床表现缺乏特异性,容易误诊,确诊主要根据病理结果。多数患者皮损较浅表,仅局限于真皮层,其组织病理改变具有特征性,多由组织细胞、淋巴细胞、嗜酸性粒细胞、泡沫细胞及典型的Touton巨细胞等浸润组成,巨细胞细胞核可成花环状排列,但早期皮损中Touton巨细胞和泡沫细胞少见或无[11-13]。免疫组化CD68阳性,而S100和CD79均阴性,但也有极少数S100阳性的报道[14,15]。本例患儿第1次活检符合本病早期病理改变,6个月后第2次活检符合本病经典型病理改变,体现了JXG皮损中组织细胞随时间推移不断成熟的过程[16]。通过皮肤镜下检查,可对JXG皮损进行分期和预后判断,同时可作为随访的一项重要检查手段[17]。JXG病程一般较长,易引起患儿家长焦虑情绪,但JXG具有自限性,在确诊本病后观察和随访是最佳策略[18],因此,对于无系统受累的患儿,应加强与家属沟通解释,尽量避免激进的侵袭性治疗,皮损多于1~5年自然消退。
