靶向雄激素受体与糖皮质激素受体的抗前列腺癌药物筛选模型的建立和应用
2020-08-14吴萌谢永丽崔香玲周金明岑山
吴萌,谢永丽,崔香玲,周金明,岑山
·论著·
靶向雄激素受体与糖皮质激素受体的抗前列腺癌药物筛选模型的建立和应用
吴萌,谢永丽,崔香玲,周金明,岑山
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所免疫室(吴萌、谢永丽、崔香玲、岑山);321004 金华,浙江师范大学化学和生命科学学院现代制药创新研究中心(周金明)
寻找可以克服由于糖皮质激素(GR)高表达造成的现有抗雄激素治疗药物耐药问题的化合物分子。
构建双荧光素酶报告系统检测模型评价化合物对雄激素受体(AR)与GR 的转录活性影响,进行基于AR 拮抗剂分子共同特征药效团模型和基于GR 蛋白晶体结构的虚拟筛选,对虚拟筛选得到的化合物分子进行初步活性评价,验证先导物分子的AR 与GR 转录抑制活性、耐药性前列腺癌细胞的增殖抑制活性。
成功构建可用来评价化合物对AR 与GR 转录活性影响情况的双荧光素酶报告系统,通过虚拟筛选与活性检测得到了化合物分子G8,G8 可以抑制AR 与GR 双靶点转录活性,抑制GR 高表达的恩杂鲁胺耐药细胞22Rv1 的增殖。
证实了通过理性药物设计与生物筛选模型筛选得到的化合物G8,可以克服由于GR 高表达造成的现有抗雄激素治疗药物耐药问题,具有潜在的临床应用价值。
雄激素受体; 糖皮质激素受体; 耐药性前列腺癌; 恩杂鲁胺
前列腺癌是西方国家男性中发病率最高的癌症类型[1]。在中国男性群体中,前列腺癌发病率近年来呈现快速增加的趋势[2]。去势治疗(androgen deprivation therapy,ADT)是晚期前列腺癌患者的常用治疗方案,但很大比例的经ADT 治疗的晚期前列腺癌患者会出现复发并最终发展成为致死性的转移性去势抵抗前列腺癌(metastatic castration-resistant prostate cancer,mCRPC),其特征为肿瘤可以通过细胞内合成的睾酮或二氢睾酮(5α-dihydrotestosterone,DHT)激活雄激素受体(androgen receptor,AR)进而生长增殖[3]。恩杂鲁胺(enzalutamide,Enza)是新一代的AR 拮抗剂,它可以通过与AR 直接结合进而拮抗其转录功能,抑制AR 下游靶基因的表达,抑制肿瘤生长,延长晚期前列腺癌患者的生存期[4-5]。
随着恩杂鲁胺在临床中的广泛应用,关于其固有或获得性耐药的报道越来越多[6-7]。有研究表明,恩杂鲁胺阻断AR 信号通路后,可以引起糖皮质激素受体(glucocorticoids receptor,GR)的高表达,高表达的GR 可以使约50% 的被恩杂鲁胺抑制的AR 调控基因重新表达,进而促进前列腺癌细胞的增殖[8]。研究显示GR 激动剂地塞米松(dexamethasone,Dex)可以导致恩杂鲁胺耐药,但GR 拮抗剂则可以使GR 高表达的前列腺癌细胞恢复对恩杂鲁胺的敏感性[9-10]。有报道称另外一种AR 信号通路抑制剂阿比特龙也可以导致前列腺癌细胞中GR 的表达量升高[9],这意味着GR 信号通路的过度激活是抗雄激素治疗中一种常见的耐药机制。因此,同时阻断AR 与GR 信号通路可能是克服在抗雄激素治疗过程中出现的GR 高表达相关耐药性问题的有效策略。
多靶点药物策略近年来日益受到研究者的关注,尤其是针对癌症、神经退行性疾病以及心血管疾病等发病机制复杂的疾病,多靶点药物拥有独特优势[11]。近期有研究者报道,吡咯咪唑聚酰胺ARE-1 可以同时抑制AR 与GR 的转录活性,并且ARE-1 可以抑制恩杂鲁胺耐药性肿瘤模型的体内增殖活性[12]。另外AR 与GR 同属于核受体超家族中的3-类固醇受体家族,两个蛋白的配体结合区具有高度的氨基酸序列相似性,尤其是在配体结合区内的荷尔蒙结合口袋区域(hormone binding pockets,HBP),两者具有高度的氨基酸序列与蛋白空间结构的相似性。因此,设计同时靶向AR 与GR 的多靶点药物,用于治疗由于GR 过表达造成的抗雄激素治疗耐药性mCRPC 具有较高可行性。
通过理性药物设计方法,结合现有抗雄激素药物的化学结构与GR 的蛋白晶体结构,我们构建了AR 与GR 双靶点拮抗剂的虚拟筛选模型与活性评价系统。从本单位“国家新药(微生物)筛选中心样品库”约4 万个小分子化合物中筛选确定了G8 为活性较好的AR 与GR 双靶点拮抗剂。化合物G8 为治疗GR 高表达耐药性mCRPC 提供了先导物。我们的工作证明了理性药物设计是开发AR 与GR 双靶点拮抗剂的有效策略。
1 材料与方法
1.1 材料
1.1.1 质粒 wt-AR 质粒由加拿大麦吉尔大学惠赠,含有pCMV 启动子,表达野生型全长AR 蛋白。PSA-luc 质粒由日本癌症化疗中心Hiroyuki 教授惠赠,共表达前列腺特异性抗原和萤火虫荧光素酶。pCMV-Renilla 质粒是本科室所有,表达海肾荧光素酶。MMTV-luc 质粒由加拿大麦吉尔大学惠赠,共表达小鼠乳癌病毒和萤火虫荧光素酶。
1.1.2 试剂
1.1.2.1 生化试剂 F-12K、RPMI1640 培养基、无酚红 RPMI1640 培养基、FBS、活性炭处理FBS、含EDTA 的0.25% 胰酶、PBS 均购自美国Gibco 公司;Lipofectamine 2000 DNA 转染试剂和MTT 试剂均购自美国Invitrogen 公司;双荧光素酶活性测定试剂盒购自美国Promega 公司。
1.1.2.2 抗体 β-actin 鼠单克隆抗体购自北京中杉金桥生物技术有限公司;GR 鼠单克隆抗体购自美国Abcam 公司。
1.1.2.3 药品 DHT 由加拿大麦吉尔大学惠赠;Enza 由上海药物所惠赠;Dex、米非司酮(mifepristone,Mif)购自美国Sigma 公司;G8 初筛来自本单位“国家新药(微生物)筛选中心样品库”,后续购买自上海陶素生化科技有限公司。
1.1.3 仪器 Centro XS3 LB 960 酶标仪购自德国Berthold 公司;EnSpire 2300 多功能酶标仪购自美国PerkinElmer 公司;凝胶成像仪Gel Doc™XR+ 购自美国Bio-Rad 公司。
1.2 方法
1.2.1 细胞培养
1.2.1.1 LNCaP 细胞 LNCaP 细胞(CRL-1740)购于ATCC,用含有10% FBS 的RPMI1640培养基进行培养,按1:2 或1:3 传代,3 ~ 4 d 传一次。
1.2.1.2 PC-3 细胞 PC-3 细胞(CRL-1435)购于ATCC,用含有10% FBS 的F-12K 培养基进行培养,按1:3 ~1:5 传代,2 ~ 3 d 传一次。
1.2.1.3 22Rv1细胞 22Rv1 细胞(CRL-2505)购于ATCC,用含有10% FBS 的RPMI1640 培养基进行培养,按1:3 ~1:5 传代,3 ~ 4 d 传一次。
1.2.2 双荧光素酶报告基因检测系统实验
1.2.2.1 检测化合物对内源性AR 的转录抑制活性 在24 孔板中,接种1.6 × 105个/ml 的LNCaP 细胞悬液,每孔接种500 μl。24 h后,每孔共转染100 ng PSA-luc 和1 ng pCMV-Renilla 质粒,转染后6 h 将培养基换成含10% 活性炭处理的FBS 的无酚红 RPMI1640 培养基;转染24 h 后,每孔按照1 nmol/L DHT 和相应药物各加入1 μl,继续培养24 h。最后将培养基吸掉,每孔加入100 μl的1 × PLB 裂解20 min,收集细胞裂解物到干净的EP 管中,离心,取上清20 μl到干净的白色96 孔板中,按照双荧光素酶报告基因检测系统试剂盒的操作指南测定荧光值。
1.2.2.2 检测化合物对外源转入AR 的转录抑制活性 在24 孔板中,接种1.4 × 105个/ml 的PC-3 细胞悬液,每孔接种500 μl。24 h 后,每孔共转染100 ng PSA-luc、20 ng wt-AR 和1 ng pCMV-Renilla 质粒,后续操作同1.2.2.1。
1.2.2.3 检测化合物对内源性GR 的转录抑制活性 在24 孔板中,接种1.4 × 105个/ml 的PC-3 细胞悬液,每孔接种500 μl。24 h 后,每孔共转染100 ng MMTV-luc 和1 ng pCMV-Renilla 质粒。转染后24 h 加药时,每孔按照100 nmol/L Dex 和相应药物各加入1 μl,后续其他操作同1.2.2.1。
1.2.3 MTT 法检测细胞存活率 取生长状态良好的22Rv1 细胞,制成2 × 104个/ml 的细胞悬液,接种于96 孔板中,每孔接种200 μl。培养24 h 后,每孔加入1 μl 相应的药物,每组药物做3 组平行,同时以DMSO 为空白对照组。继续培养72 h,每孔加入20 μl 用PBS 配制的5 g/L 的MTT 溶液,继续培养4 h 后,吸弃上清,每孔用100 μl 异丙醇溶解沉淀物,96 孔板振荡器上混匀20 min,最后用EnSpire 2300 多功能酶标仪在570 nm 波长下测定其吸光度值。
1.2.4 免疫印迹(Western blot) 接种3 × 105个/ml的细胞悬液于6 孔板中,培养72 h,吸弃培养基,加入80 μl 预冷RIPA(强)裂解液,刮取6 孔板中细胞,收集到1.5 ml EP 管中。冰浴裂解30 min,每隔10 min 振荡混匀一次。加入20 μl 5 × 蛋白上样缓冲液,金属浴煮30 min 制样,10% SDS-PAGE 分离蛋白样品。70 V 恒压湿法转膜80 min 后(采用PVDF 膜),5% 脱脂奶粉室温封闭1 h。先后与一抗和二抗孵育,然后ECL 显色。抗体使用浓度为GR(1:1000),β-actin(1:5000),山羊抗兔(1:5000),山羊抗小鼠(1:5000)。
1.2.5 虚拟筛选 使用Discovery Studio 3.5 软件中的“prepare ligands”模块将本单位“国家新药(微生物)筛选中心样品库”中的化合物分子生成三维结构,使用本实验室之前构建的AR 拮抗剂分子共同特征药效团模型Pharmacophore_8[13],利用Discovery Studio 3.5 进行基于药效团模型的虚拟筛选,取Fitvalue 大于3 的化合物分子。从PDB 网站(www.pdb.org)下载GR 蛋白晶体结构(PDB 编码:4MDD),以其HBP 位点自带配体为中心、12 nm 为半径的球体作为对接位点,使用GOLD 软件对接,取分值排名在前300 的分子,进一步人工挑选出结构新颖的候选分子进行活性初筛。
1.3 统计学处理
采用GraphPad Prism 5.01 软件对数据进行分析,两组间数据比较采用检验,以0.05为具有统计学显著性差异。
2 结果
2.1 AR 与 GR 转录活性检测模型的构建
AR 可被DHT 激活,进而进入细胞核触发一系列的下游反应,PSA 是AR 调控的关键下游靶基因之一,因此检测PSA 的表达量可用以评价AR 的转录活性,MMTV 则可以用来评价细胞内GR 的转录活性,基于此,首先构建了可以评价AR 和GR 转录活性的细胞检测模型。PC-3 细胞是AR 阴性的前列腺癌细胞系,我们的实验结果显示该细胞可以大量表达GR,LNCaP 是AR 阳性细胞系,细胞内不表达GR,而对恩杂鲁胺耐药的AR 阳性细胞系22Rv1 细胞则可以表达GR(图1A)。在PC-3 中转入AR 质粒使其可以表达野生型AR 蛋白,图1B 实验结果显示,与空白组相比,DHT 可以激活AR 转录活性,从而提高PSA表达水平约3.8 倍,而作为阳性对照的第二代AR 拮抗剂恩杂鲁胺则可以抑制AR 的转录活性,使得PSA 表达量下降至空白组水平。在AR 阳性的LNCaP 细胞中,我们观察到了同样的现象(图1C),以上两组实验证明该实验体系可用来评价化合物对AR 转录活性的影响。PC-3 细胞是GR 阳性前列腺癌细胞系,MMTV 可以被GR 激活从而表达量上调,图1D 实验结果显示,GR 激动剂地塞米松激活GR 转录活性后使得MMTV 表达量相较于空白组升高约6.6 倍,而在阳性药GR 拮抗剂米非司酮处理组中,GR 转录活性被几乎100% 的抑制,证明该实验体系可用来评价化合物对GR 转录活性的影响。
2.2 虚拟筛选与先导物的确定
利用目前文献报道或已上市的6 个AR 拮抗剂的分子结构,我们实验室前期构建了AR 拮抗剂分子共同特征药效团模型Pharmacophore_8。利用该模型,对本单位“国家新药(微生物)筛选中心样品库”中40618 个化合物分子首先进行了基于药效团模型的虚拟筛选,得到了909 个匹配度大于3 的化合物分子,随后利用GOLD 对接软件,对909 个分子进行了基于GR 蛋白晶体结构(PDB 编号:4MDD)的对接,对打分排名前300 的化合物,利用可视化筛选最终挑选了165 个结构新颖的分子进行活性初筛。初筛结果显示化合物G8(图 2A)具有潜在的AR 与GR 双靶点拮抗活性,且G8 的分子结构与Pharmacophore_8 匹配度较高(图2B)。
2.3 G8 具有 AR 与 GR 双靶点转录抑制活性
为了进一步评价化合物G8 对AR 与GR 的双靶点拮抗水平,我们利用双荧光素酶报告系统首先检测了不同浓度的G8 对AR 的转录抑制活性,图3A显示,在PC-3 细胞中,G8 可以剂量依赖性地抑制外源性AR 转录水平,同样的,G8 对于内源性AR 也显示出了较好的剂量依赖性抑制活性(图3B),在20 μmol/L 浓度处理下,G8 对外源与内源性AR 的抑制水平均接近100%,且G8 在5 μmol/L 浓度下仍对外源与内源性AR有显著抑制。图3C显示,G8 可以剂量依赖性地抑制内源性GR 的转录水平,在20 μmol/L 浓度处理下,G8 对内源性GR 的转录抑制率约为61.3%。上述实验结果表明,G8 可以同时抑制AR 与GR 的转录活性,从而抑制这两个信号通路的功能。
2.4 G8 可以抑制 GR 高表达的恩杂鲁胺耐药性前列腺癌细胞增殖活性
为了确定G8 是否可以克服由于GR 高表达造成的恩杂鲁胺耐药性问题,我们利用MTT 实验检测了G8 对GR 高表达的恩杂鲁胺耐药性AR阳性前列腺癌细胞22Rv1 的增殖抑制活性。图4显示,恩杂鲁胺对22Rv1 细胞的增殖抑制活性很弱(IC50> 50 μmol/L),但G8 却可以抑制22Rv1 细胞的增殖活性(IC50= 10.2 μmol/L)。结果显示,通过同时阻断AR 与GR 的转录活性,G8 对于GR 高表达造成的恩杂鲁胺耐药性前列腺癌仍具有增殖抑制活性。
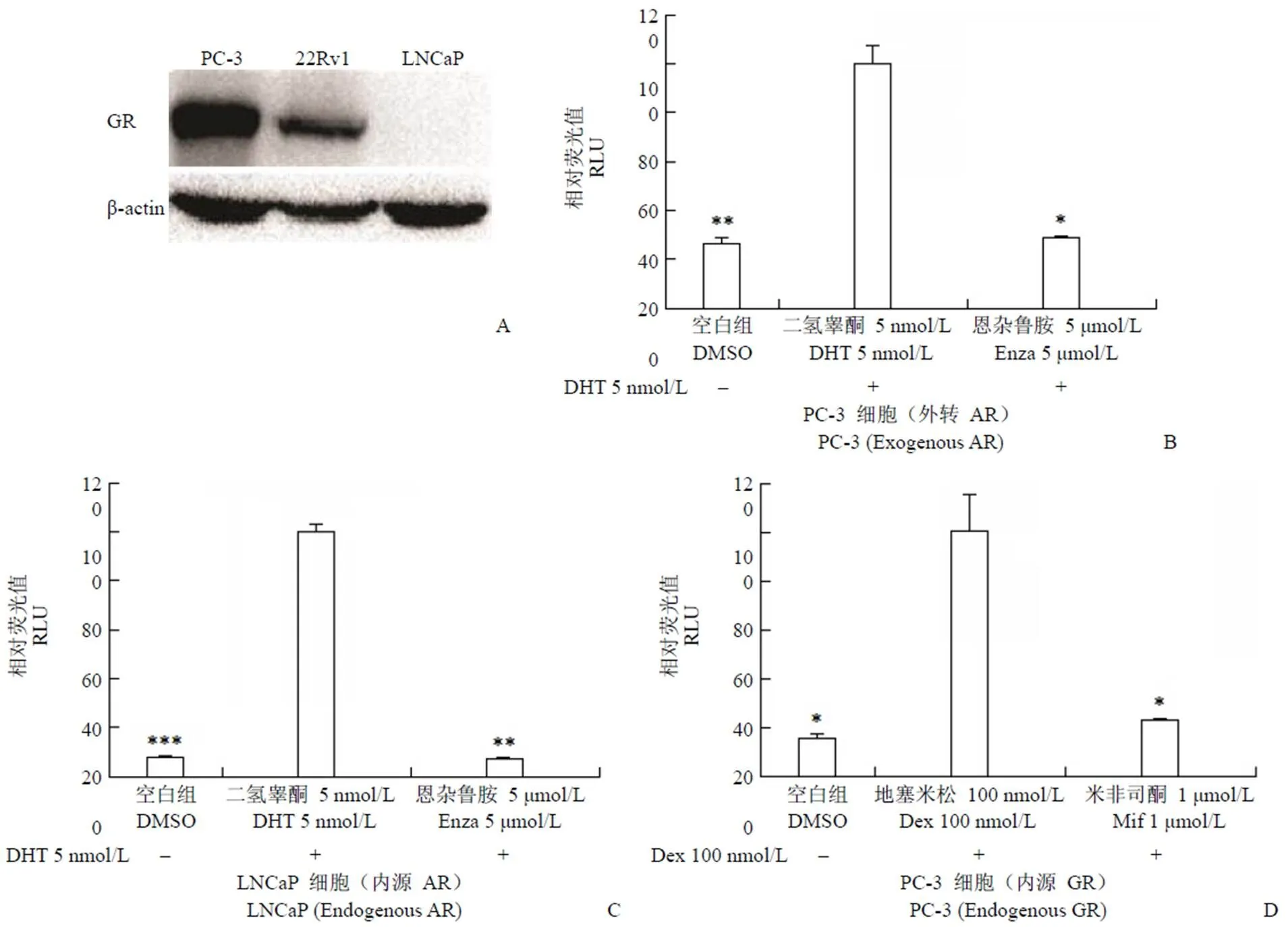
图 1 AR 与GR 转录活性评价模型的构建(A:Western blot 检测不同前列腺癌细胞系中GR 的表达水平;B:检测化合物对外转AR 转录活性影响的双荧光素酶报告系统;C:检测化合物对内源性AR 转录活性影响的双荧光素酶报告系统;D:检测化合物对内源性GR 转录活性影响的双荧光素酶报告系统;*P < 0.05,**P < 0.01,***P < 0.001)
Figure 1 Construction of AR and GR transcription activity evaluation models (A: GR expression levels in different prostrate cancer cells detected by Western blot; B: Dual-luciferase reporter system that test the exogenous AR transcription inhibition activity;C: Dual-luciferase reporter system that test the endogenous AR transcription inhibition activity; D: Dual-luciferase reporter system that test the endogenous GR transcription inhibition activity;*0.05,**0.01,***0.001)
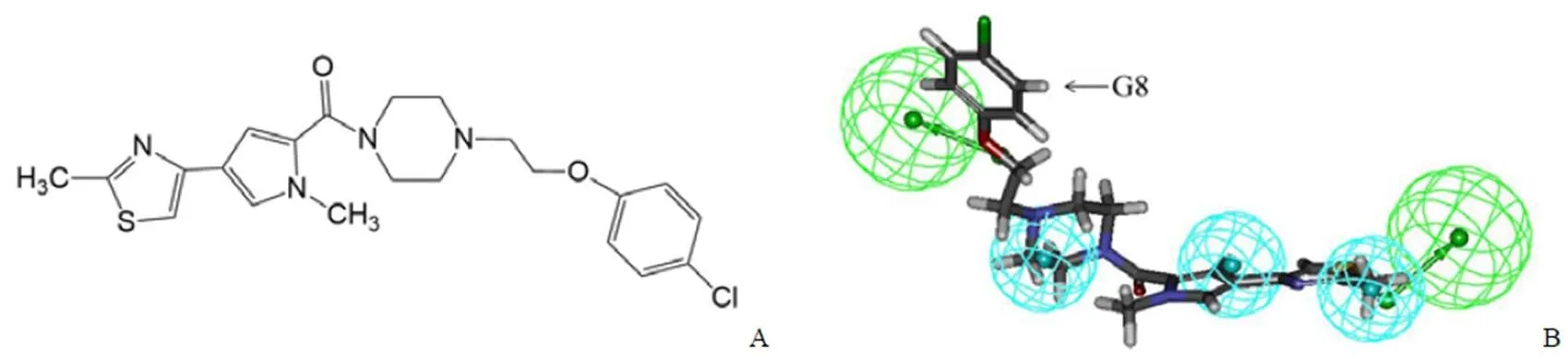
图 2 AR 与GR 双靶点拮抗剂G8 的分子结构(A)与药效团模型_8 的匹配度(B)
Figure 2 Structure of AR and GR dual antagonist G8 (A) and its matched degree of pharmacophore_8 (B)
3 讨论
晚期去势抵抗前列腺癌患者在接受抗雄激素治疗过程中,耐药性问题一直是制约患者生存期的重要因素。为了克服由于GR 高表达造成的抗雄激素治疗耐药性问题,我们通过理性药物设计方法,首先利用目前已有的AR 拮抗剂分子,构建了基于分子共同特征的药效团模型,随后我们开展了基于AR 拮抗剂分子共同特征药效团模型的虚拟筛选以及基于GR 蛋白结构的对接筛选,从筛选得到的分子中,我们挑选了部分结构较新颖的分子进行了活性初筛,初步确定了化合物G8 为候选分子。随后的实验证明,通过同时阻断AR 与GR 的转录活性,G8 可以抑制GR 高表达造成的恩杂鲁胺耐药性前列腺癌细胞的增殖活性,为后续开发AR 与GR 双靶点拮抗剂治疗耐药性前列腺癌提供了结构先导物。
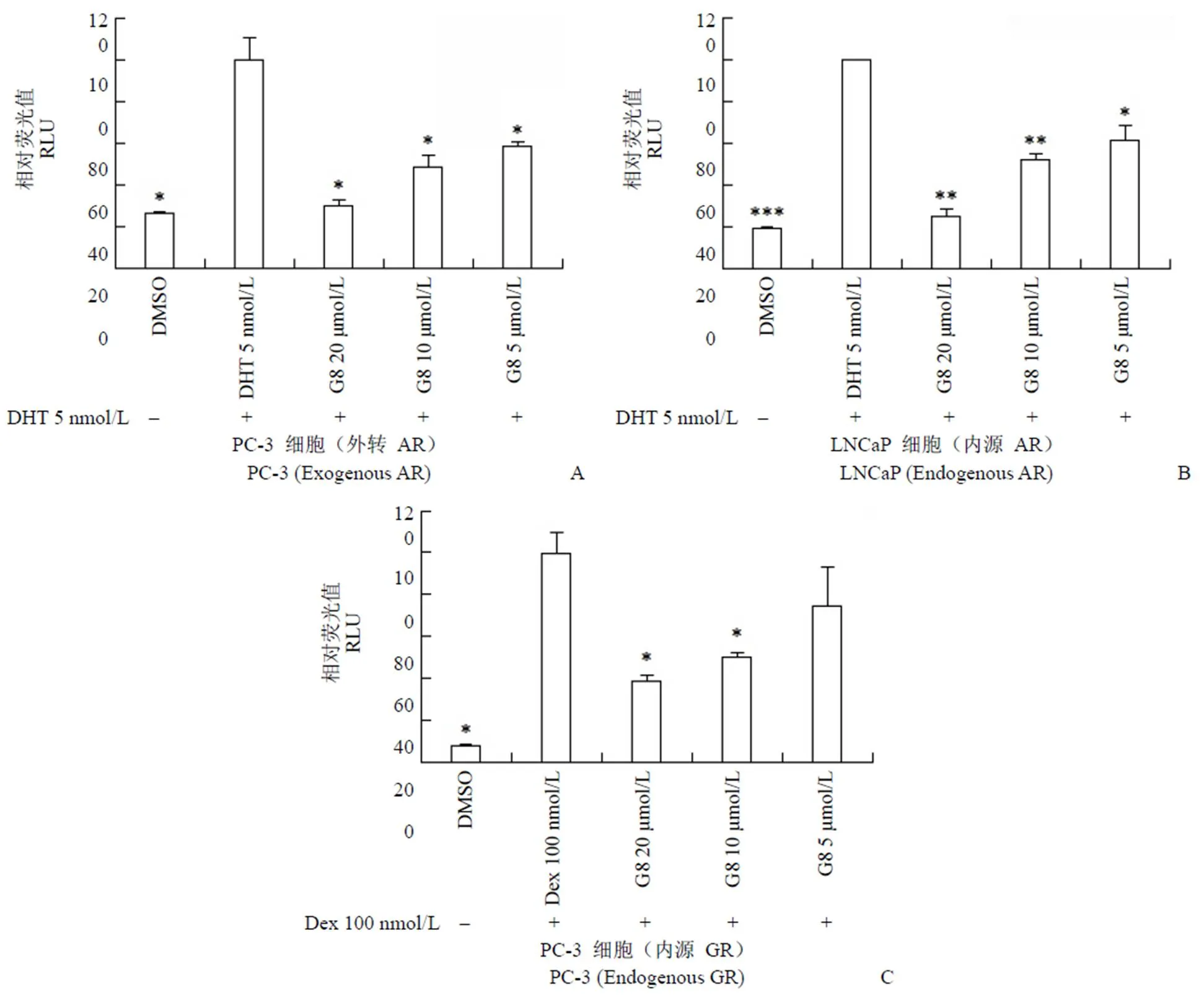
图 3 不同浓度G8 对AR 与GR 的转录抑制活性(A:G8 对外源性AR 的转录抑制活性;B:G8 对内源性AR 的转录抑制活性;C:G8 对内源性GR 的转录抑制活性;*P < 0.05,**P < 0.01,***P < 0.001)
Figure 3 AR and GR transcription inhibition activity of different concentration of G8 (A: Exogenous AR transcription inhibition activity of G8; B: Endogenous AR transcription inhibition activity of G8; C: Endogenous GR transcription inhibition activity of G8;*0.05,**0.01,***0.001)
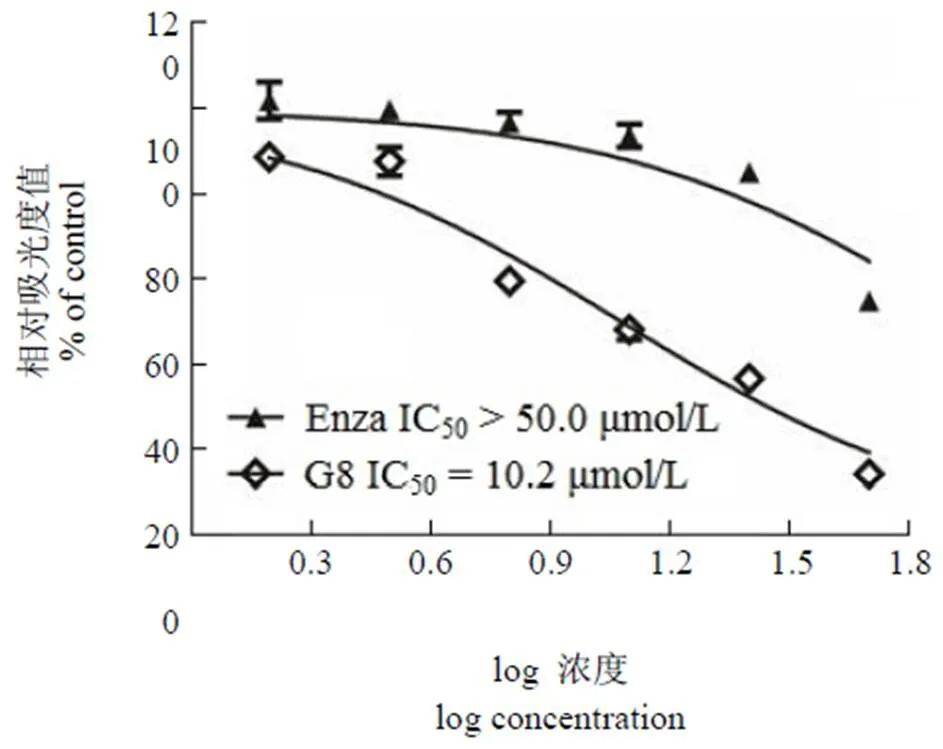
图 4 G8 对去势抵抗前列腺癌细胞 22Rv1 细胞的增殖抑制活性
Figure 4 The 22Rv1 cells proliferation inhibition activity of G8
多靶点药物成为新药开发越来越重要的方向之一,例如阿比特龙通过作用于CYP17、3β-HSD 以及AR 来发挥抗前列腺癌活性。但值得注意的是,研发同时特异性靶向两个甚至多个靶点的药物在实际操作中具有更高的难度。我们的研究证明,通过综合利用靶点与配体的信息,联合药效团模型虚拟筛选以及基于蛋白结构的虚拟筛选的理性药物设计方法,在开发多靶点药物领域具有重要的应用价值。
[1] Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin, 2018, 68(6):394-424.
[2] Liu X, Yu C, Bi Y, et al. Trends and age-period-cohort effect on incidence and mortality of prostate cancer from 1990 to 2017 in China. Public Health, 2019, 172:70-80.
[3] Fujita K, Nonomura N. Role of androgen receptor in prostate cancer: a review. World J Mens Health, 2019, 37(3):288-295.
[4] Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med, 2012, 367(13): 1187-1197.
[5] Davis ID, Martin AJ, Stockler MR, et al. Enzalutamide with standard first-line therapy in metastatic prostate cancer. N Engl J Med, 2019, 381(2):121-131.
[6] Claessens F, Helsen C, Prekovic S, et al. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat Rev Urol, 2014, 11(12):712-716.
[7] Culig Z. Molecular mechanisms of enzalutamide resistance in prostate cancer. Curr Mol Bio Rep, 2017, 3(4):230-235.
[8] Arora VK, Schenkein E, Murali R, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell, 2013, 155(6):1309-1322.
[9] Isikbay M, Otto K, Kregel S, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer, 2014, 5(2):72-89.
[10] Puhr M, Hoefer J, Eigentler A, et al. The glucocorticoid receptor is a key player for prostate cancer cell survival and a target for improved antiandrogen therapy. Clin Cancer Res, 2018, 24(4):927-938.
[11] Ravikumar B, Aittokallio T. Improving the efficacy-safety balance of polypharmacology in multi-target drug discovery. Expert Opin Drug Discov, 2018, 13(2):179-192.
[12] Kurmis AA, Yang F, Welch TR, et al. A pyrrole-imidazole polyamide is active against enzalutamide-resistant prostate cancer. Cancer Res, 2017, 77(9):2207-2212.
[13] Wu M, Xie Y, Cui X, et al. Rational drug design for androgen receptor and glucocorticoids receptor dual antagonist. Eur J Med Chem, 2019, 166:232-242.
Construction and application of a drug screening model for anti-prostate cancer agents targeting androgen receptor and glucocorticoids receptor
WU Meng, XIE Yong-li, CUI Xiang-ling, ZHOU Jin-ming, CEN Shan
Laboratory of Immunology, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China (WU Meng, XIE Yong-li, CUI Xiang-ling, CEN Shan); Drug Discovery & Innovation Center, College of Chemistry and Life Sciences, Zhejiang Normal University, Jinhua 321004, China (ZHOU Jin-ming)
To find agents that could overcome drug resistance of antiandrogens caused by GR over expression.
In silico screening was performed to screen our compounds database, dual-luciferase reporter systems were used to evaluate the influence of compounds on AR and GR transcription activity, 22Rv1 cell proliferation inhibition activity of the lead compound G8 was detected by MTT assay.
The AR and GR transcription inhibition activity evaluation model was constructed successfully. Compound G8 screened by our model could inhibit AR and GR transcription activity and inhibit 22Rv1 cell proliferation activity.
This study demonstrates that compound G8 screened through rational drug design and bioactivity evaluation, overcomes drug resistance of antiandrogens caused by GR over expression.
Androgen receptor; Glucocorticoids receptor; Drug resistance prostate cancer; Enzalutamide
ZHOU Jin-ming, Email: zhoujinming@zjnu.edu.cn; CEN Shan, Email: shancen@hotmail.com
国家自然科学基金(81672559)
周金明,Email:zhoujinming@zjnu.edu.cn;岑山,Email:shancen@imb.pumc.edu.cn
10.3969/j.issn.1673-713X.2020.04.006
2020-01-07
