利用生物信息学分析筛选乳腺癌不良预后的关键基因
2020-08-14曹林韩丽杨明韩彬刘福
曹林,韩丽,杨明,韩彬,刘福
·论著·
利用生物信息学分析筛选乳腺癌不良预后的关键基因
曹林,韩丽,杨明,韩彬,刘福
637000 南充,川北医学院附属医院药剂科
利用生物信息学方法,通过分析基因表达数据库(GEO)基因芯片数据筛选与乳腺癌不良预后相关的核心基因,为乳腺癌的治疗提供新的候选靶点。
从 GEO 数据库下载微阵列数据集 GSE15852,采用 GEO 在线工具 GEO2R 筛选差异表达基因(DEGs);DAVID 数据库对筛选出的差异表达基因,进行基因本体论分析(GO)和京都基因与基因组百科全书(KEGG)通路富集分析;基于 STRING 数据库和 Cytoscape 软件构建蛋白质-蛋白质相互作用关系(PPI),并用Cytoscape 软件 MCODE 插件进行模块分析,获取关键基因;用在线工具 Kaplan-Meier Plotter 对这些关键基因进行生存分析,获取与乳腺癌预后不良的相关核心基因;采用基因表达谱交互分析(GEPIA)进一步验证。
筛选出 57 个差异表达基因,其中上调基因 17 个,下调基因 40 个。上调基因主要富集在雌激素反应、对细胞运动的负调控反应、心脏右心室形态发生、交感神经系统发育、细胞-细胞黏附及输尿管的萌芽发育等生物过程;聚焦于造血细胞系信号通路。下调基因显著富集在脂质代谢、分解、存储过程,胆固醇的储存、运输,甘油三酯的合成分解代谢,血管生成等生物过程;聚焦于 PPAR 信号通路、对脂肪细胞脂肪分解的调节作用、脂肪细胞因子信号通路等途径。PPI 网络及 MCODE 模块分析鉴定出 7 个核心基因,关键基因的生存分析及 GEPIA 分析发现24 和基因的高表达患者生存率低于低表达患者。
该方法为寻找乳腺癌不良预后的关键基因、探索乳腺癌治疗新靶点提供一定依据。
乳腺癌; 生物信息学; GEO 数据库; 差异表达基因; 关键基因
乳腺癌是女性发病率最高的恶性肿瘤之一,是全球女性癌症死亡的主要原因。在全球范围内,2018 年有大约 210 万新诊断的女性乳腺癌病例,占女性癌症病例的近四分之一[1]。随着筛查方法及治疗手段的发展,乳腺癌患者的生存率有所提高。但有研究报道,I 期确诊的乳腺癌患者 5 年相对生存率接近 100%,而对于那些被诊断为 IV 期乳腺癌的患者 5 年相对生存率下降到 26%,揭示乳腺癌的晚期患者生存率较低[2]。手术、化疗和放疗等传统治疗方法对晚期乳腺癌患者来说并不能提供理想的治疗结果[3]。此外,乳腺癌肿瘤的异质性,使乳腺癌的治疗结果个体差异大。乳腺癌发病机制复杂,目前对其潜在的分子机制尚不完全清楚。因此,迫切需要探索更特异、更经济的生物标志物来预测乳腺癌的预后,开发更好的治疗策略和更好地了解其潜在机制的靶点。
近年来,基于高通量平台的微阵列已成为筛选癌症发生过程中重要的遗传或表观遗传学改变及寻找癌症诊断和预后的有前途的生物标志物的有效工具[4]。生物信息学分析基于基因芯片,通过数据筛选、统计分析、可视化手段、分子互作网络和通路分析等方法整合海量、复杂的生物学信息,挖掘潜在的生物标记物,为疾病的治疗提供新的策略。
鉴于乳腺癌在女性的高发病率和死亡率,不少学者进行乳腺癌研究。早期诊断和分子靶向治疗迫切需要确定决定乳腺癌进展、转移和不良预后的关键基因。Li 等[5]研究表明,LAPTM4B、VEGF 和核 survivin 的表达与乳腺癌患者的各种临床病理特征和预后显著相关,可以被视为乳腺癌的治疗靶点。文献研究报道一些与乳腺癌不良预后分子生物标记物,如 KPNA2 有助于关键蛋白的异常定位和乳腺癌的预后不良[6],RASSF1A 甲基化对女性乳腺癌预后不良的预测作用[7]等。本研究通过分析基因表达数据库(gene expression omnibus,GEO)中基因芯片,旨在探讨乳腺癌不良预后的关键差异基因,希望能获得更多与乳腺癌预后相关的分子机制的生物学信息,为治疗乳腺癌提供新的靶点。
1 资料与方法
1.1 微阵列数据集下载
美国国立生物技术中心(NCBI)的 GEO 数据库是一个免费的基因表达公共数据库(https:// www.ncbi.nlm.nih.gov/geoprofiles/),从 GEO 下载乳腺癌的微阵列数据集 GSE15852。芯片信息:[HG-U133A] Affymetrix Human Genome U133A Array,ID:20097481,平台:GPL96。该数据集包含 43 例乳腺癌组织和 43 例正常乳腺组织基因表达数据。
1.2 方法
1.2.1 差异表达基因分析 用GEO2R在线工具筛选乳腺癌标本与正常乳腺标本之间的差异表达基因(differentially expressed genes,DEGs),筛选条件|logFC| > 2(FC 为差异倍数),矫正后值 < 0.01。logFC < 0 的 DEGs 为下调基因,log FC > 0 的 DEGs 为上调基因。
1.2.2 基因本体论和京都基因与基因组百科全书通路富集分析 DAVID 是一个在线生物信息学工具,用于基因/蛋白质功能注释和功能基因集富集。将筛选的 DEGs 输入 DAVID6.8(https://david. ncifcrf.gov)进行基因本体论(gene ontology analysis,GO)功能注释,包括生物过程(biological processes,BP)、分子功能(molecular function,MF)、细胞成分(cell component,CC);京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)通路富集分析,寻找差异表达基因富集的关键信号通路。以< 0.05 认为差异具有统计学意义。
1.2.3 蛋白质-蛋白质相互作用关系网络和模块分析 采用 STRING11.0 数据库(https://string-db. org/cgi/input.pl),设置置信度为 0.04,构建乳腺癌差异表达基因蛋白质-蛋白质相互作用关系网络(protein-protein interaction networks,PPI),用Cytoscape 3.7.2 软件进行对 PPI 网络可视化,并用 Cytoscape 软件的 MCODE 插件进行模块分析,筛选关键基因。
1.2.4 生存分析 通过在线数据库 Kaplan-Meier Plotter(https://kmplot.com/analysis/)中乳腺癌样本的生存率,对“1.2.3”中 MCODE 筛选出的关键基因进行总生存(overall survival,OS)分析。logrank< 0.05 被认为具有统计学意义。筛选出乳腺癌患者生存率较差的基因,采用基因表达谱交互分析(gene expression profiling interactive analysis,GEPIA)进一步验证,获取与乳腺癌不良预后的关键基因。
2 结果
2.1 差异基因表达
GEO 在线工具GEO2R 基因芯片 GSE15852 进行分析,以 |logFC| > 2 和矫正后< 0.01 为标准,筛选出 57 个差异表达基因,其中上调基因17 个,下调基因 40 个(表 1)。
2.2 GO 功能富集分析和 KEGG 通路富集分析
通过 DAVID 网站对 57 个差异表达基因进行 GO 功能富集分析和 KEGG 通路富集分析,GO 分析包括生物过程(BP)、细胞成分(CC)和分子功能(MF)。结果表明,上调基因显著富集在雌激素反应、对细胞运动的负调控反应、心脏右心室形态发生、交感神经系统发育、细胞-细胞黏附及输尿管的萌芽发育等生物过程;主要聚焦于造血细胞系信号通路。而下调基因显著富集在脂质代谢、分解、存储过程,胆固醇的储存、转运,甘油三酯的合成分解代谢,血管生成等生物过程;聚焦于 PPAR 信号通路、对脂肪细胞脂肪分解的调节作用、脂肪细胞因子信号通路等途径(图 1 和表 2)。
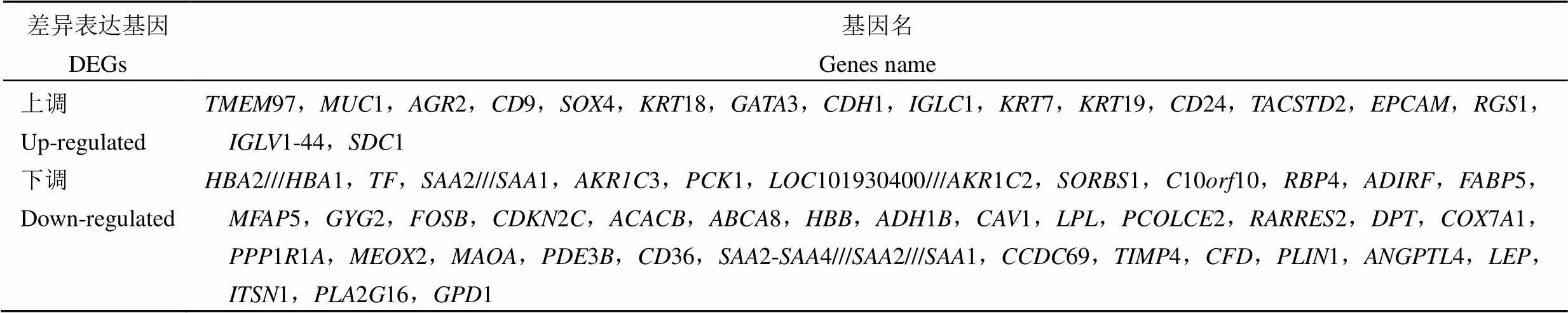
表 1 差异表达基因(17 个上调基因和40 个下调基因)(P < 0.01)
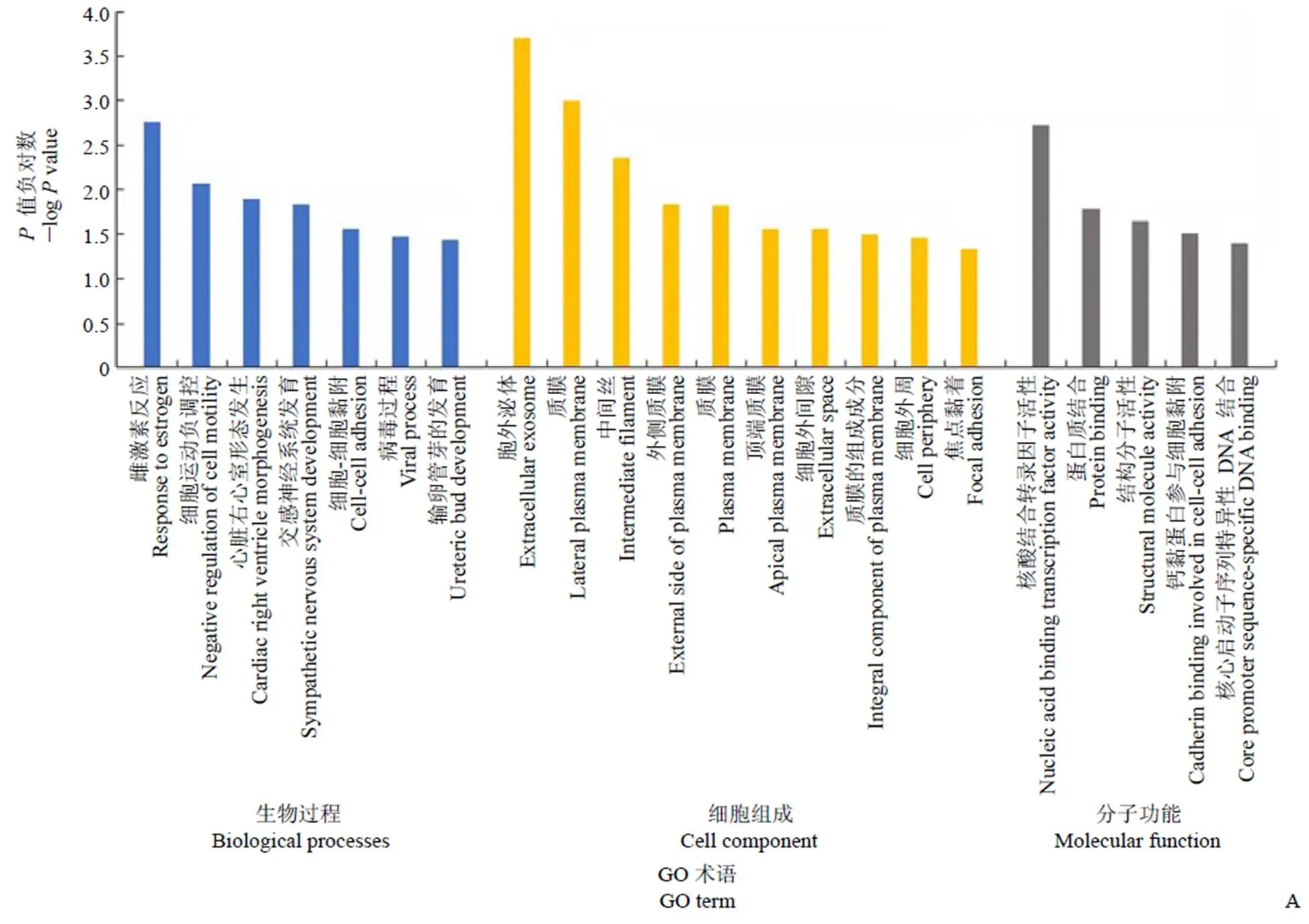
Figure 1 Genes ontology enrichment analysis of differentially expressed genes (A: Up-regulated genes; B: Down-regulated genes;< 0.05)

表 2 差异表达基因 KEGG 通路富集分析(P < 0.05)

图 2 差异表达基因的PPI 网络和模块分析(A:差异表达基因的PPI 网络可视化结果;B:关键模块;红色为上调基因,蓝色为下调基因,连接线表示差异表达基因之间的相互作用)
Figure 2 DEGs PPI network complex and the module analysis (A:Differentially expressed gene protein interaction network visualization results; B: Key module; Up-regulated genes were marked in red, down-regulated genes were marked in blue, and the lines show the interaction between the DEGs)
2.3 PPI 网络和模块分析
STRING11.0 在线数据库构建 PPI 网络。57 个差异表达基因中共有 37 个差异表达基因(14 个上调基因和 23 个下调基因)被过滤到 PPI 网络复合体中,该复合体包括 37 个节点和 86 条边,而 57 个 DEG 中有 20 个没有过滤到 PPI 网络复合体中(图 2A)。然后,通过 Cytotype MCODE 插件识别关键模块。以 Node Score Cutoff = 0.2,K-Core = 2,Max. Depth = 100 为标准,在 37 个节点中共鉴定出 7 个中心节点,均为上调基因(图 2B)。
2.4 生存分析
7 个核心候选基因的预后信息可在免费的在线 Kaplan-Meier 绘图仪数据库中获得。结果发现,3 个基因的总体存活率明显较差,而 4 个基因的总体存活率无显著性差异(< 0.05,表 3 和图 3)。随后,利用 GEPIA 网站进一步验证了总体存活率有显著差异的 3 个基因在癌症患者和正常人之间的表达状况。结果表明,2 个基因(24 和)在乳腺癌()组织中的表达高于正常乳腺组织(< 0.01,图 4)。高表达患者的生存率比低表达患者的更差。

表 3 7 个核心基因的预后信息
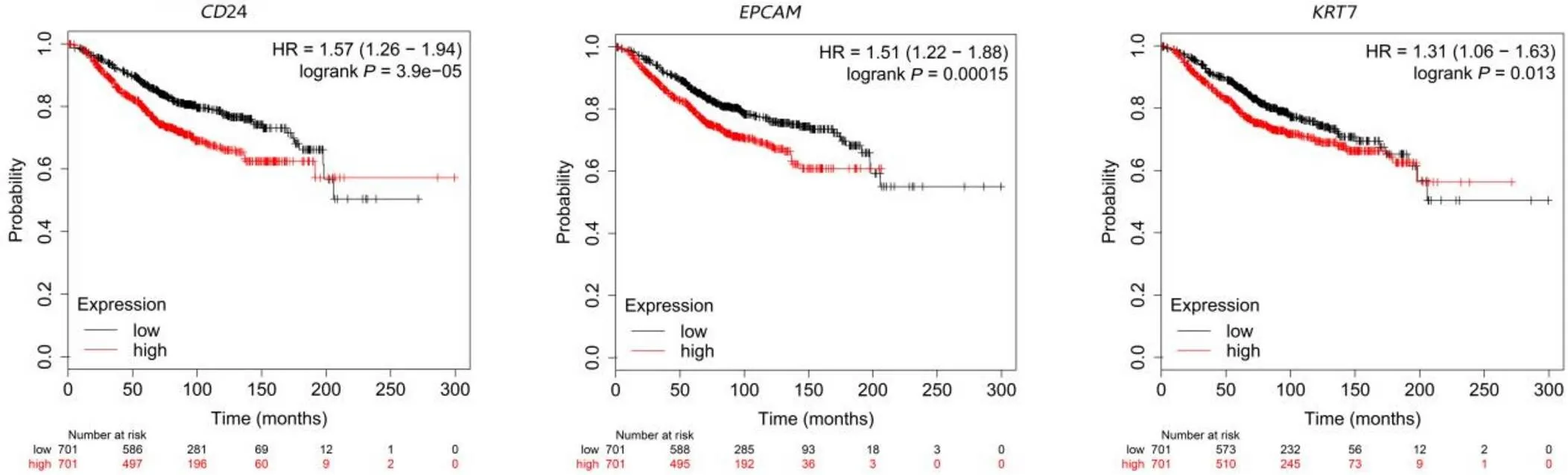
图 3 核心基因的Kaplan-Meier 预后价值(Logrank P < 0.05 认为有统计学意义)
Figure 3 Prognostic values of the key genes by Kaplan-Meier Plotter (Logrank< 0.05 was considered statistically significant)
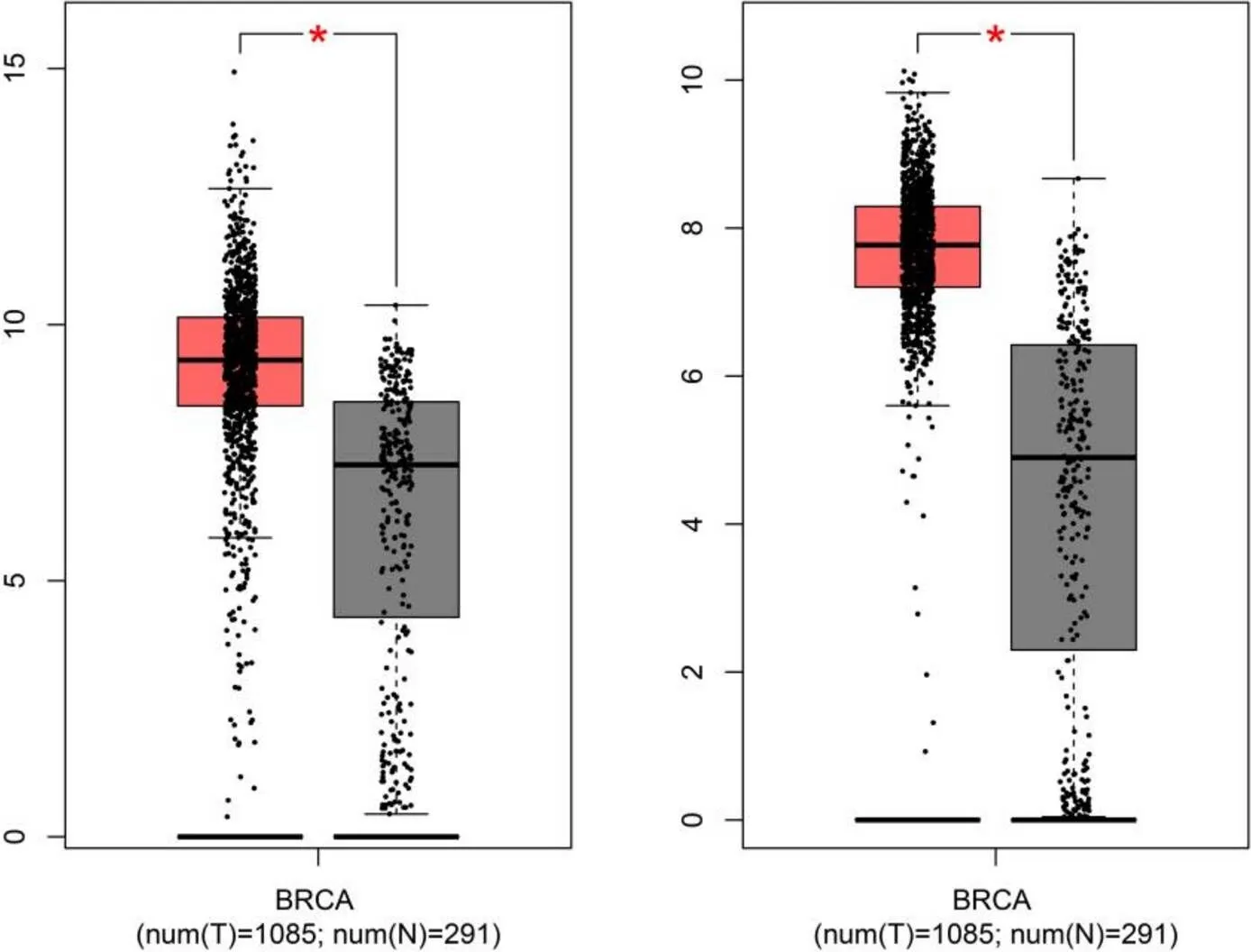
图 4 基因表达谱交互分析进一步验证核心基因CD24(A)和EPCAM(B)在BRCA 标本中的表达水平,并与正常标本进行对照(红框表示癌组织组,灰色表示正常组织组,*P < 0.01;点表示每个样本的表达)
Figure 4 Gene expression profiling interactive analysis was performed to further demonstrate the genes' expression level of core genes24 (A) and(B) in BRCA samples contrasted to normal samples (Red box means the cancer tissue group, gray means the normal tissue group,*< 0.01. The dots represented expression in each sample)
3 讨论
乳腺癌是一种异质性疾病,根据雌激素受体(ER)、孕激素受体(PR)、人表皮生长因子受体2(HEGFR 2)和增殖标志物 Ki67(MKI67)[8-10]的表达可分为四大分子亚型。目前,乳腺癌患者的预后预测主要基于这一分类和常规的临床病理特征,如组织学分级、组织学类型和 TNM 分期。然而,在临床实践中,肿瘤的异质性给治疗效果和预后的预测带来了极大的困难。近年来,乳腺癌生物学过程中涉及的分子机制研究取得了很大进展。随着新批准的基因治疗策略的出现[11],增加的癌基因正在被测试为癌症的治疗靶点。基因的变化已经被确认在乳腺癌的发生和发展中起着关键作用[12-15]。因此,需要进一步探索更有效的分子生物标记物用于乳腺癌的预防、诊断和治疗。
本研究利用 GEO 数据库的基因芯片 GSE15852 进行生物信息学分析乳腺癌组织与正常乳腺组织的差异基因,鉴定出 57 个差异表达基因,其中上调基因 17 个,下调基因 40 个。通过 GO 分析和 KEGG 通路分析,发现主要富集在细胞运动和脂质代谢等通路。文献报道脂质对癌症的影响已被广泛研究,对不同的癌症有不同的影响[16]。通过 PPI 网络及 MCODE 挖掘出 7 个核心基因,进一步进行生存分析和 GEPIA 分析,发现 CD24 和 EPCAM 基因在癌症中的表达高于正常组织,患者的生存率更低。CD24 是糖基-磷脂酰-肌醇连接的糖蛋白,在包括癌细胞在内的多种细胞类型中表达。文献表明,CD24 在多种肿瘤发生和发展中起作用,包括肺癌、前列腺癌、卵巢癌等[17]。已经有报道表明24 的过表达对癌细胞中突变的 p53 蛋白的失活至关重要[18]。本研究们结果表明,24 是乳腺癌治疗的关键基因,可以作为一种很有前途的治疗靶点和预后标志物,与文献[19-20]报道一致。我们试图确定细胞内24 的位置,并确定位置是否影响肿瘤表型和患者预后,以便最终允许开发最优的24 定向治疗。EPCAM 是一种跨膜糖蛋白,其过度表达被认为与不同肿瘤的增殖增强和恶性程度有关[21]。Baccelli 等[22]和Sadeghi 等[23]报道,的过表达提高了乳腺癌患者的转移率,提示EPCAM 可能是一个潜在的恶性肿瘤的生物标志物,与本文结果一致。是乳腺癌中重要的过表达基因,可作为预后因素进行评估。
综上所述,本研究利用 GEO 数据集结合生物信息学的综合分析,发现 2 个核心基因与 BRCA 的进展和预后相关,可能会为 BRCA 潜在的生物标志物和生物学机制提供一些有用的信息和方向,为制定有效的诊断和治疗策略提供参考。
[1] Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. A Cancer J Clin, 2018, 68(6):394-424.
[2] Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. A Cancer J Clin, 2019, 69(5):363-385.
[3] Zheng S, Li M, Miao K, et al. SNHG1 contributes to proliferation and invasion by regulating miR-382 in breast cancer. Cancer Manag Res, 2019, 11:5589-5598.
[4] Song E, Song W, Ren M, et al. Identification of potential crucial genes associated with carcinogenesis of clear cell renal cell carcinoma.J Cell Biochem, 2018, 119(7):5163-5174.
[5] Li S, Wang L, Meng Y, et al. Increased levels of LAPTM4B, VEGF and survivin are correlated with tumor progression and poor prognosis in breast cancer patients. Oncotarget, 2017, 8(25):41282-41293.
[6] Alshareeda AT, Negm OH, Green AR, et al. KPNA2 is a nuclear export protein that contributes to aberrant localisation of key proteins and poor prognosis of breast cancer. Br J Cancer, 2015, 112(12):1929- 1937.
[7] Buhmeida A, Merdad A, Al-Maghrabi J, et al. RASSF1A methylation is predictive of poor prognosis in female breast cancer in a background of overall low methylation frequency. Anticancer Res, 2011, 31(9):2975-2981.
[8] Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A, 2003, 100(14):8418-8423.
[9] Sørlie T, Wang Y, Xiao C, et al. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics, 2006, 7:127.
[10] Goldhirsch A, Wood WC, Coates AS, et al. Strategies for subtypes-dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol, 2011, 22(8):1736-1747.
[11] Stein CA, Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol Ther, 2017, 25(5):1069-1075.
[12] Buys SS, Sandbach JF, Gammon A, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer, 2017, 123(10):1721-1730.
[13] Li G, Guo X, Tang L, et al. Analysis of BRCA1/2 mutation spectrum and prevalence in unselected Chinese breast cancer patients by next-generation sequencing. J Cancer Res Clin Oncol, 2017, 143(10): 2011-2024.
[14] Cybulski C, Carrot-Zhang J, Kluźniak W, et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet, 2015, 47(6):643-646.
[15] Dan X, Pu C, Mengjiao H, et al. An integrated bioinformatical analysis to evaluate the role of KIF4A as a prognostic biomarker for breast cancer. Onco Targets Ther, 2018, 11:4755-4768.
[16] Zhang J, Zhou YJ, Yu ZH, et al. Identification of core genes and clinical roles in pregnancy-associated breast cancer based on integrated analysis of different microarray profile datasets. Biosci Rep, 2019, 39(6):BSR20190019.
[17] Lee JH, Kim SH, Lee ES, et al. CD24 overexpression in cancer development and progression: a meta-analysis. Oncol Rep, 2009, 22(5):1149-1156.
[18] Wang L, Liu R, Ye P, et al. Intracellular CD24 disrupts the ARF–NPM interaction and enables mutational and viral oncogene-mediated p53 inactivation. Nat Commun, 2015, 6:5909-5919.
[19] Suyama K, Onishi H, Imaizumi A, et al. CD24 suppresses malignant phenotype by downregulation of SHH transcription through STAT1 inhibition in breast cancer cells. Cancer Lett, 2016, 374(1):44-53.
[20] Zhang P, Zheng P, Liu Y. Amplification of the CD24 gene is an independent predictor for poor prognosis of breast cancer.Front Genet, 2019, 10:560.
[21] van der Gun BT, Melchers LJ, Ruiters MHJ, et al. EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis, 2010, 31(11):1913-1921.
[22] Baccelli I, Schneeweiss A, Riethdorf S, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol, 2013, 31(6):539-544.
[23] Sadeghi S, Hojati Z, Tabatabaeian H. Cooverexpression of EpCAM and c-myc genes in malignant breast tumours. J Genet, 2017, 96(1): 109-118.
Identification of key genes with poor prognosis in breast cancer by bioinformatical analysis
CAO Lin, HAN Li, YANG Ming, HAN Bin, LIU Fu
Department of Pharmacy, Affiliated Hospital of North Sichuan Medical College, Nanchong 637000, China
Bioinformatic method was used to analyze the gene expression database (GEO) gene chip data to screen core genes related to poor prognosis of breast cancer and to provide a new candidate target for the treatment of breast cancer.
Microarray dataset from GEO database GSE15852 was downloaded and differentially expressed genes (DEGs) were screened using GEO online tool GEO2R. Next, Gene Ontology and Kyoto Encyclopedia of Gene and Genome pathway enrichment analyses were performed using the online databases DAVID for selected DEGs. Protein-protein interaction relationship (PPI) was constructed based on STRING database and Cytoscape software, and the key genes were obtained by module analysis with MCODE plug-in of Cytoscape software. Then, overall survival analysis was performed using the Kaplan–Meier curve to screen core genes related to the prognosis of breast cancer, and the genes were further validated in gene expression profiling interactive analysis (GEPIA).
57 DEGs were identified in BRCA in the dataset, including 17 up-regulated genes largely enriched in the response to estrogen, negative regulation of cell motility, cardiac right ventricle morphogenesis, sympathetic nervous system development, cell-cell adhesion, viral process, ureteric bud development biological processes and hematopoietic cell lineage signaling pathway, and 40 down-regulated genes specifically enriched in lipid metabolic process, lipid transport, lipid storage, positive regulation of cholesterol storage, cholesterol transport, triglyceride catabolic process, angiogenesis biological processes, PPAR signaling pathway, regulation of lipolysis in adipocytes, and adipocytokine signaling pathway. By extracting key modules from the PPI network by Cytotype MCODE plugin, all 7 up-regulated genes were selected. In addition, survival analysis and gene expression profiling interactive analysis of key genes showed that the survival rate of patients with high expression of24 andgenes was lower than that of patients with low expression.
This study provides a basis for finding the key genes with poor prognosis of breast cancer and exploring new targets for the treatment of breast cancer.
Breast cancer; Bioinformatics; GEO database; Differentially expressed genes; Key genes
LIU Fu, Email: nclf91@163.com
四川省教育厅项目(18ZB0219)
刘福,Email:nclf91@163.com
10.3969/j.issn.1673-713X.2020.04.012
2020-02-26
