CuRh2O4热力学稳定性研究
2020-08-12吴泳洁戴炳蔚周晓龙
吴泳洁,王 鑫,戴炳蔚,周晓龙,于 杰
(昆明理工大学材料科学与工程学院,昆明 650093)
0 引 言
Rh在化学周期表中位于Co的正下方,其化学性质与Co相近。在化学结构上,Rh的氧化物和Co的氧化物可以合成相似的层状结构[1],因此,与典型的Co基氧化物热电材料相比,铑基氧化物材料被认为是一类重要的新型热电材料。其中Cu-Rh-O三元氧化物在催化、电化学等领域有潜在的应用。Murthy等[2]发现CuRh2O4导电性能优良,且在温度高于290 ℃时有更好的电导率。Kurita等[3]通过扩展Heikes公式发现,Rh3+在低自旋状态可以得到更大的ZT值。Shibasaki等[1]发现在Rh基三元氧化物中掺杂Zn、Mg元素可以提高电子输运效率,改善热电性能。Usui等[4]通过第一性原理方法发现空穴对LaRhO3和CuRhO2热电性能会产生影响。Toyoda等[5]通过密度泛函理论(DFT)计算发现Cu-X-O(X为过渡金属)三元氧化物具有高的催化活性,这与过渡金属X的外层电子结构有关。Hinogami等[6]发现铜铑铜铁矿(CuRhO2)具有优异的伏安循环特性,可以作为电解水的高活性阳极。Jacob[7]通过设计固态电池研究了CuRh2O4和CuRhO2的反应温度,得到了电化学反应过程中CuRh2O4和CuRhO2的反应吉布斯形成能。综上,Cu-Rh-O三元氧化物在实际生产中具有广泛的潜在应用价值,而对于采用传统粉末烧结法制备这类材料时,铑氧化物价格昂贵,无法进行大量的制备研究。通过第一性原理下的热力学计算,可以得到反应过程中各项物质的热力学性质。根据热力学性质可以评估反应体系中的氧化物稳定性,研究反应方向在热力学上的反应趋势,尤其在反应热力学计算的过程中,热力学计算得到的结果可以补充数据手册中缺失的热力学数据。
本文在验证计算可靠性的基础上,通过热力学计算反应过程,并且通过第一性原理模拟计算研究CuRh2O4热力学稳定性,判断CuO+Rh2O3→CuRh2O4反应在热力学条件的趋势,旨在为CuRh2O4的合成和类似材料的制备提供热力学理论依据。
1 实 验
1.1 实验仪器
所用仪器设备:FA2004N电子天平,玛瑙研磨罐,YP-1400压片机,GXL-25高温箱式炉,戴尔PowerEdge T630塔式服务器任务调度软件为Torque,版本号4.2.10[8]。
1.2 实验过程
1.2.1 第一性原理方法计算CuRh2O4热力学性质
采用Materials Studio 8.0[9]软件的CASTEP程序包计算CuRh2O4热力学性质。首先对晶胞模型进行几何优化,然后对优化成功后的晶胞模型进行热力学性质计算。计算精度设置为超精细(Utra-fine)。收敛误差精度为:能量在5.0×10-6eV/atom以内,最大作用力为0.1 eV/nm,最大应力为0.02 GPa,最大原子位移距离为5.0×10-5nm。电子交换相关函数[10]利用广义梯度近似GGA的PBE泛函。最大迭代次数为9999。计算赝势采用超软赝势(Ultrasoft Pseudopotentials)和模守恒赝势(Norm Conserving Pseudopotentials)两种分别计算,超软赝势计算时,截断能统一为440 V,使用模守恒赝势计算时截断能统一为880 V。k点精度设置为精细。SCF自洽循环收敛精度为1.0×10-10eV/atom。
1.2.2 固相烧结法制备CuRh2O4圆片
按照摩尔比1∶1的比例称取CuO(纯度>99.9%)和Rh2O3(纯度>99.9%)粉末,充分研磨后在24 MPa的压力下压片,放置箱式炉中进行固相反应烧结。温度为600 ℃、880 ℃、900 ℃、940 ℃、970 ℃和1 000 ℃,保温72 h,随炉冷却至室温。
1.2.3 分析测试
热失重-差热分析(TG-DTA)采用上海精密科学仪器有限公司的 ZRY-2P 型高温热重分析仪及CRY-2P型高温差热分析仪,量程为±100 μV,升温速率为10 K·min-1,测量温度范围为25~1 100 ℃。物相检测采用XRD测试,仪器型号为D/max-2200X,管电压40 kV,管电流40 mA,扫描范围5°~90°。
2 结果与讨论
Dollase和O′Neill[11]在1997年测得常温常压下的CuRh2O4的晶体结构,空间点群为I41/amd,晶格常数为a=b=0.617 56 nm,c=0.900 60 nm,α=β=γ=90°。通过XRD检测,确定了Rh2O3和CuO的晶体结构。Rh2O3的晶体结构是Biesterbos和Hornstra[12]在1973年测得的,空间点群为Pbca,晶格常数为a=0.514 77 nm,b=0.544 25 nm,c=1.469 77 nm,α=β=γ=90°。CuO的晶体结构是Asbrink和Waskowska[13]在1991年测得的,其空间点群为C1/c1,晶格常数为a=0.469 27 nm,b=0.342 83 nm,c=0.513 7 nm,α=γ=90°,β=99.546°。本文中计算出的不同相的晶体结构模型如图1所示。

图1 晶体结构模型Fig.1 Crystal structure model
通过MS软件中CASTEP模块计算了两种不同赝势下的结构优化值,部分值与实验值进行对比,如表1所示。超软赝势下的结构与实验值的晶格参数更加接近,且超软赝势下能量比模守恒的能量低,因此超软赝势下的结构更加稳定。在后期计算过程中采用超软赝势进行计算。

表1 实验值和计算值的晶胞参数及能量对比表Table 1 Comparison of unit cell parameters and energy between experimental and calculated values
图2为CuO和Rh2O3的热力学参数对比图。如图2所示,根据声子谱结果得到CuO和Rh2O3的相对焓(ΔH)、熵×温度(T×S)、相对Gibbs自由能(ΔG)随温度变化的函数曲线,与《实用无机物热力学数据手册》[14]中CuO和Rh2O3在25 ℃至1 027 ℃的熵、焓、自由能随温度变化的热力学数据相吻合。验证了计算数据具有准确性。CASTEP计算中振动自由能是通过准谐波近似(QHA)获得的,该近似方法仅在晶体熔点以下才具有较高的精度。温度越高,熵焓和自由能的误差就越大[15],因此图2中的数据在高温段偏差较大。由于CuRh2O4相在《实用无机物热力学数据手册》中没有相应热力学数据,在以上计算数据验证可靠性的前提下,图3为通过CASTEP计算得到的热力学数据。
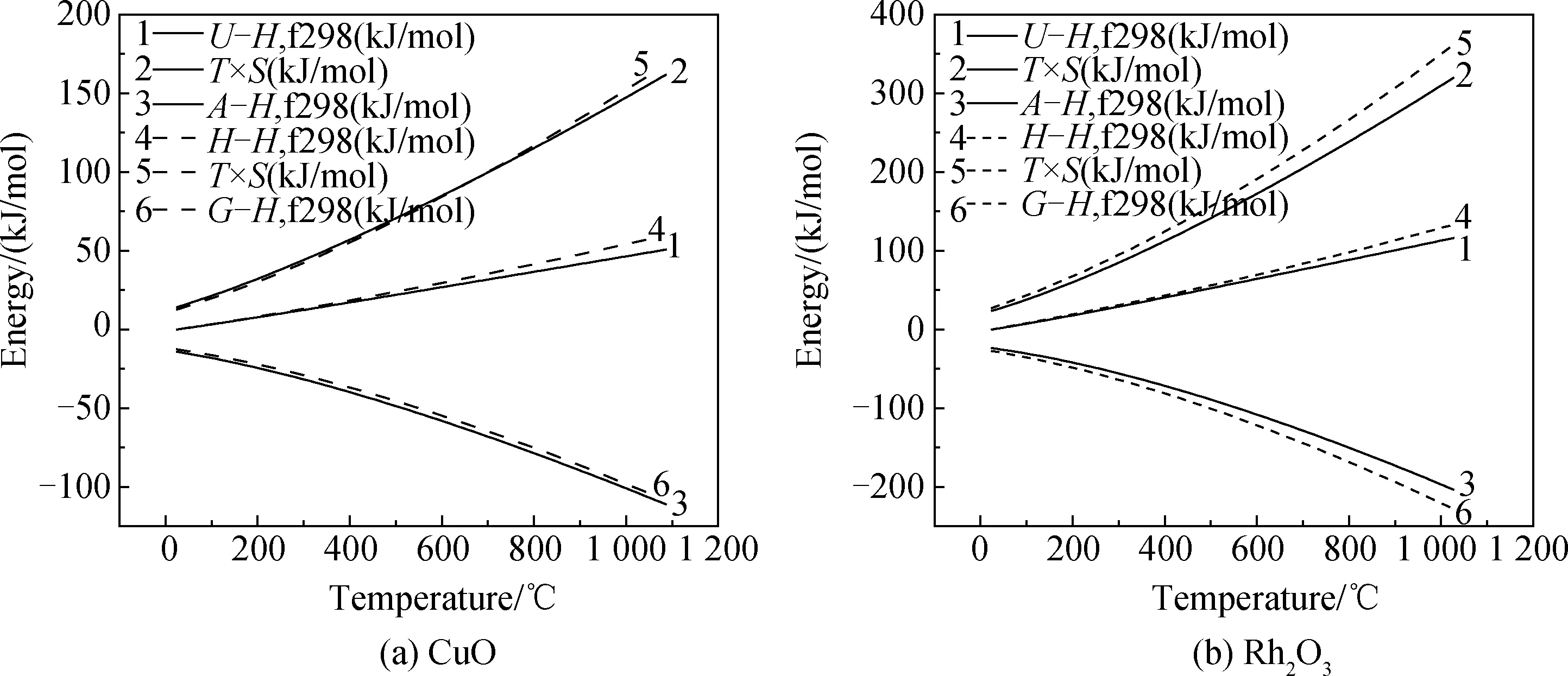
图2 CuO和Rh2O3的热力学参数对比图Fig.2 Comparison of thermodynamic parameters of CuO and Rh2O3

图3 CuRh2O4的热力学参数Fig.3 Thermodynamic parameters of CuRh2O4
图4是CuRh2O4、CuO和Rh2O3的T×S-温度函数曲线图,对于CuO+Rh2O3→CuRh2O4相变反应中所涉及到的三种物质,CuRh2O4的T×S值曲线最大,其次是Rh2O3,最小的是CuO。也就是说对于这三种物质来说,CuRh2O4是最趋于稳定的,而CuO是最不稳定的,Rh2O3的稳定性介于CuRh2O4和CuO之间。根据实验反应可知,CuRh2O4是最趋于稳定的,CuRh2O4的稳定性比CuO和Rh2O3的稳定性好。这验证了计算的准确性。

图4 CuO、Rh2O3和CuRh2O4的T×S温度函数Fig.4 T×S function of CuO, Rh2O3 and CuRh2O4
利用CASTEP计算了反应中CuO、Rh2O3和CuRh2O4的声子态度,由于固体中的赫姆霍兹自由能近似的等价吉布斯自由能,式(1)为反应式,利用计算所得的生成物(CuRh2O4)的赫姆霍兹自由能减去反应物(CuO和Rh2O3)的赫姆霍兹自由能,得到其反应Gibbs自由能差,如式(2)所示。
CuO+Rh2O3→CuRh2O4
(1)
ΔrG(T)=G298(T)(CuRh2O4)-G298(T)(CuO)-G298(T)(Rh2O3)
(2)
其中,ΔrG(T)为反应Gibbs自由能,G298(T)M(M:CuRh2O4、CuO、Rh2O3)为各反应物相的Gibbs自由能。
CuO和Rh2O3反应的Gibbs自由能随温度变化曲线计算结果如图5所示,在25 ℃至1 174 ℃温度区间内,Gibbs自由能的值均小于0。根据自由能反应判据可知,该反应在热力学研究下可以自由发生,在600 ℃得到最小值且反应的热力学驱动力最大,当温度大于1 174 ℃时,该反应在热力学上ΔrG(T)>0,表明该反应不能发生。

图5 反应Gibbs自由能变化Fig.5 ΔrG(T) of the reaction
为了确定固相反应CuO+Rh2O3→CuRh2O4能否进行及反应温度,进一步验证计算的准确性。通过差热分析实验得到了CuO和Rh2O3(摩尔比1∶1)的混合粉末在25~1 200 ℃下的吸-放热量变化,如图6所示,由图可知,900 ℃存在反应的吸热峰,991 ℃时达到极值,说明反应及反应温度与计算结果一致。910 ℃开始质量变化,说明在此温度下伴随发生了其他反应。

图6 CuO和Rh2O3差热分析图(25~1 200 ℃)Fig.6 Differential thermal analysis of CuO and Rh2O3(25-1 200 ℃)
如图7(a)所示,样品的XRD谱表明,当保温温度为600 ℃(保温72 h)时,为Rh2O3衍射峰,没有出现CuRh2O4,表明此温度下反应变化不明显,即在热力学计算驱动力最大的情况下,反应发生受到了动力学因素的制约,同时与CuO和Rh2O3粉体之间互扩散驱动力弱有关。当保温温度为880 ℃(保温72 h)时,开始出现CuRh2O4和Rh2O3的混合峰。当保温温度为900 ℃(保温72 h)条件下,完全生成了CuRh2O4,如图7(b)所示。CuRh2O4的标准PDF卡片与该温度下的衍射峰完全吻合,且无杂峰,证明在900 ℃温度下CuO和Rh2O3完全生成CuRh2O4。随着温度升高,当保温温度为940 ℃时,除了CuRh2O4之外,还出现了CuRhO2,但此时仍以CuRh2O4为主。继续提高保温温度,当达到970 ℃时,仍然是CuRhO2和CuRh2O4的混合相,但此时CuRhO2远多于CuRh2O4。当保温温度为1 000 ℃时,此时完全生成了CuRhO2。可以得出,CuO+Rh2O3→CuRh2O4反应发生的温度区间为880~970 ℃。前文中计算结果表明,温度高于1 174 ℃后,Gibbs自由能大于0,该反应不再发生,对比实验结果可知,实验上限温度与计算上限误差相差174 ℃。
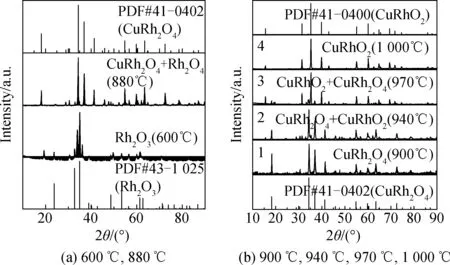
图7 CuRh2O4的XRD谱Fig.7 XRD patterns of CuRh2O4
当保温温度为1 000 ℃时,为CuRh2O4纯相衍射峰,说明在生成CuRh2O4的同时,吸热发生了相转变生成CuRhO2,在相转变过程中有O2的生成从而导致了质量减小。此外,对于CuO+Rh2O3→CuRh2O4反应,该反应属于放热反应,但由于CuRh2O4吸热发生了相转变生成CuRhO2,使得原本的放热反应被覆盖。观察940 ℃XRD谱可知,在940 ℃时出现了少量的CuRhO2,因此质量损失也同样说明CuRh2O4相转变生成CuRhO2的过程中有O2的生成从而导致了质量减小[16]。
3 结 论
(1)通过第一性原理声子谱计算得到声子谱热力学计算值与手册计算值吻合,验证了计算结果的可靠性。对比三个物相的T×S随温度变化曲线得到CuRh2O4的稳定性优于CuO和Rh2O3。
(2)反应Gibbs自由能计算结果表明,存在CuO+Rh2O3→CuRh2O4的反应,热力学上,CuO+Rh2O3→CuRh2O4反应在25~1 174 ℃区间内Gibbs自由能小于0,该反应在热力学上有发生反应的趋势,温度高于1 174 ℃后,Gibbs自由能大于0,该反应不再发生。
(3)固相反应烧结实验表明,温度在880 ℃下生成了CuRh2O4,900 ℃能够合成纯相的CuRh2O4,此反应实验区间为880~970 ℃。提高反应温度至1 000 ℃,CuRh2O4在高温下不稳定发生相变生成了CuRhO2,实验上限温度与计算上限误差相差174 ℃。
