抑制PDK1经由ASK1/JNK/Bim通路诱导慢性粒细胞白血病细胞凋亡
2020-07-21惠凌云王亚文
王 方,王 昕,刘 哲,惠凌云,冯 艾,李 娜,王亚文,
1.西安交通大学第一附属医院检验科,陕西 西安 710061;
2.西安交通大学第一附属医院生物样本资源中心,陕西 西安 710061
慢性粒细胞白血病(chronic myelogenous leukemia,CML)是起源于造血干细胞的恶性增殖性疾病[1]。95%的患者骨髓细胞携带t (9,22)染色体易位。该突变形成的BCR/ABL融合基因编码并表达BCR/ABL融合蛋白。作为非受体型酪氨酸激酶,BCR/ABL融合蛋白在细胞质中能够自身磷酸化活化并激活下游STAT5、PI3K及Ras等信号通路,影响细胞的正常生物学行为[2-4]。以伊马替尼(imatinib)为代表的特异性酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI)能够靶向抑制BCR/ABL基因的磷酸化活化,并为CML的治疗带来了革命性突破[5]。然而也有诸多局限,首先,急变期CML易获得二次突变,TKI对于该期患者疗效欠佳[6];其次,即使获得缓解的患者也存在复发的可能,而TKI并不能很好地控制复发病情;再次,近年来大量研究证实,CML肿瘤干细胞的存活并不依赖于BCR/ABL,即使被TKI完全阻断,肿瘤干细胞也不能有效地被诱导凋亡[7]。因此,寻找替代治疗靶点是治愈CML亟待解决的问题。本研究通过分析CML患者及健康体检者标本发现,3-磷酸肌醇依赖性蛋白激酶1(3-phosphoinositide-dependent kinase-1,PDK1)在CML细胞中表达升高。抑制其活性对肿瘤细胞具有明显的抗增殖和促凋亡的效应,同时也能够增强细胞对imatinib的敏感性。因此作为潜在的治疗靶点,PDK1值得后续深入研究。
1 材料和方法
1.1 主要试剂
PDK1小分子抑制剂GSK2334470购自美国Sigma-Aldrich公司,imatinib购自美国Selleck公司,细胞计数试剂盒(cell counting kit-8,CCK-8)及Annexin Ⅴ-FITC/PI凋亡试剂盒购自杭州联科生物技术股份有限公司,外周血单个核细胞分离试剂盒购自天津市灏洋生物制品科技有限责任公司,RPMI-1640培养基购自美国HyClone公司,胎牛血清购自以色列BI公司,PDK1、磷酸化PDK1、凋亡信号调节激酶1(apoptosis signalregulating kinase 1,ASK1)、磷酸化ASK1、c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)、磷酸化JNK、caspase-3及多聚ADP核糖聚合酶[poly (ADP-ribose)polymerase,PARP]单克隆抗体均购自美国Cell Signaling Technology公司,蛋白激酶B(protein kinase B,AKT)、磷酸化AKT、糖原合成酶激酶-3β(glycogen synthase kinase-3β,GSK-3β)、磷酸化GSK-3β、β-catenin、磷酸化β-catenin、Bcl-2相互作用细胞凋亡介导因子(Bcl-2 interacting mediator of cell death,Bim)、磷酸化Bim及c-Myc抗体均购自美国Santa Cruz公司,GAPDH抗体购自杭州贤至生物科技有限公司,HRP标记的羊抗鼠、羊抗兔免疫球蛋白G(immunoglobulin G,IgG)购自美国Santa Cruz公司。
1.2 细胞培养
K562和KU812细胞购自北京北纳创联生物技术研究院,用含10%胎牛血清的RPMI-1640培养基,置于37 ℃、CO2体积分数为5%的培养箱中培养,收集对数生长期的细胞用于后续实验。
1.3 克隆形成实验
收集对数生长期细胞,用终浓度为5 μmol/L的GSK2334470处理,12 h后按约500个细胞/孔的密度加入含0.9%甲基纤维素的24孔板中培养,平行3次,1周后在光学显微镜下计数克隆细胞数。
1.4 CCK-8检测
按5000个细胞/孔的密度接种于96孔板中,总体积200 μL,处理组为5 μmol/L的GSK2334470,对照组为相应浓度的二甲基亚砜(dimethyl sulfoxide,DMSO)溶剂(<0.1%)。每组设置4个平行对照孔。分别于培养24、48、72 h后加入20 μL CCK-8溶液。37 ℃培养30 min后检测450 nm处的吸光度(D)值,并比较组间差异。
1.5 蛋白质印迹法(Western blot)检测
细胞经相应处理48 h后收集,临床标本中外周血单个核细胞分离采用密度梯度离心法(依照试剂说明书),最终收集的细胞经预冷PBS洗涤3次。加入含1%苯甲基磺酰氟(phenylmethanesulfonyl fluoride,PMSF)的RIPA裂解液 (106个细胞/100 μL),冰上裂解30 min;4 ℃,13000 rpm离心15 min,吸取上清液。通过B C A 法检测蛋白浓度,加入相应体积的上样缓冲液,沸水浴中煮5 min,分装,-80 ℃保存备用。采用变性十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDSPAGE)分离蛋白质,浓缩胶和分离胶聚丙烯酰胺浓度分别为6%和10%~12%,每组上样20~50 μg。120 V恒压电泳2 h。横流湿转法转膜,40 mA转6~8 h,于5%牛血清蛋白(bovine serum albumin,BSA)中室温封闭1 h。4 ℃温育一抗过夜,二抗室温温育1~2 h,化学发光法检测信号。
1.6 细胞凋亡检测
细胞处理同Western blot,收集细胞,预冷PBS洗3次,用结合缓冲液重悬细胞,浓度约为106/mL,取100 μL细胞悬液,加入Annexin-Ⅴ/PI各5 μL室温避光染色30 min,上机检测。
1.7 统计学处理
本研究各项实验均重复3次以上,应用SPSS 20.0软件对结果进行统计学分析,资料以x±s表示。采用LSD-t法检验。P<0.05为差异有统计学意义。
2 结果
2.1 PDK1在CML标本中表达升高
检测8例初发CML患者外周血单个核细胞中PDK1蛋白水平,并与同期健康体检者相比较,PDK1在CML患者中表达明显升高(P<0.05,图1)。
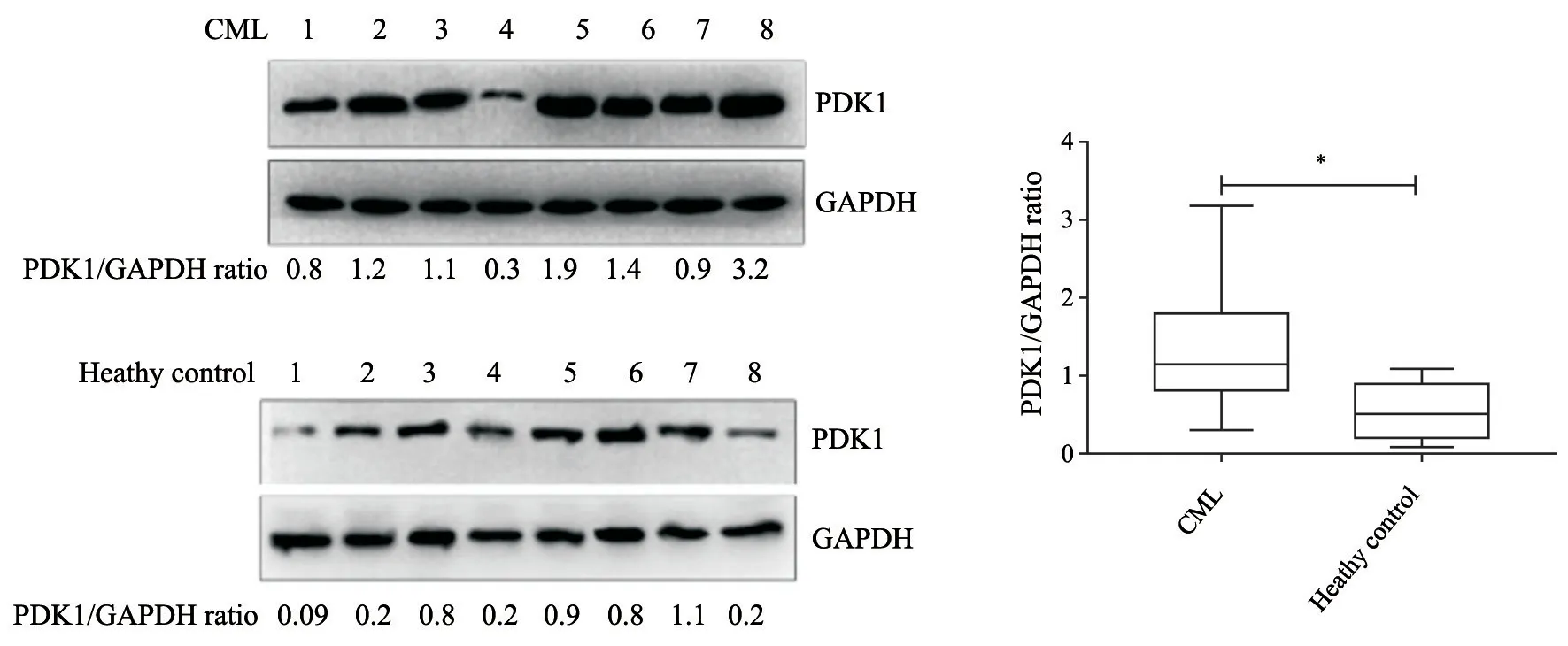
图1 Western blot法检测8例CML患者及8例健康体检者外周血中单个核细胞PDK1表达丰度Fig.1 PDK1 abundance was detected by Western blot in peripheral blood mononuclear cells from 8 newly diagnosed CML patients and 8heathy donors
2.2 抑制PDK1影响细胞增殖
在K562和KU812两株CML细胞中,PDK1小分子抑制剂GSK2334470能够明显抑制细胞增殖。克隆形成实验显示,相比对照组,抑制剂组细胞克隆数目明显降低(K562:P<0.01;KU812:P<0.05),且形态较对照组小,细胞较为分散(图2A)。另外,CCK-8检测结果也显示,在处理后72 h,抑制剂组细胞活力明显低于对照组(K562:P<0.01;KU812:P<0.001,图2B)。
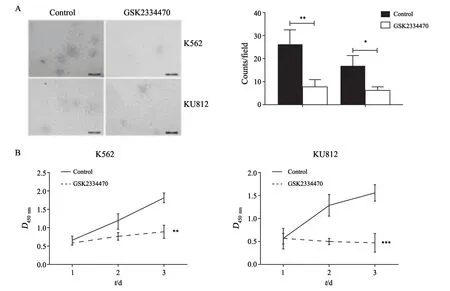
图2 克隆形成实验(A)和CCK-8(B)检测GSK2334470对细胞增殖的抑制作用Fig.2 Inhibition of cell growth by GSK2334470 was detected by clone formation assay (A) and CCK-8 assay (B)
2.3 抑制PDK1促进细胞凋亡
流式细胞术分析结果显示,处理组细胞凋亡率明显提高,K 562 细胞中,凋亡率为19.2%±4.38%(P<0.01),而KU812细胞则更为敏感,凋亡率为35.7%±6.33%(P<0.01,图3A)。随后检测了凋亡信号通路中执行分子caspase-3及底物PARP的活化片段,结果显示,GSK2334470能够明显诱导两者活化(图3B)。
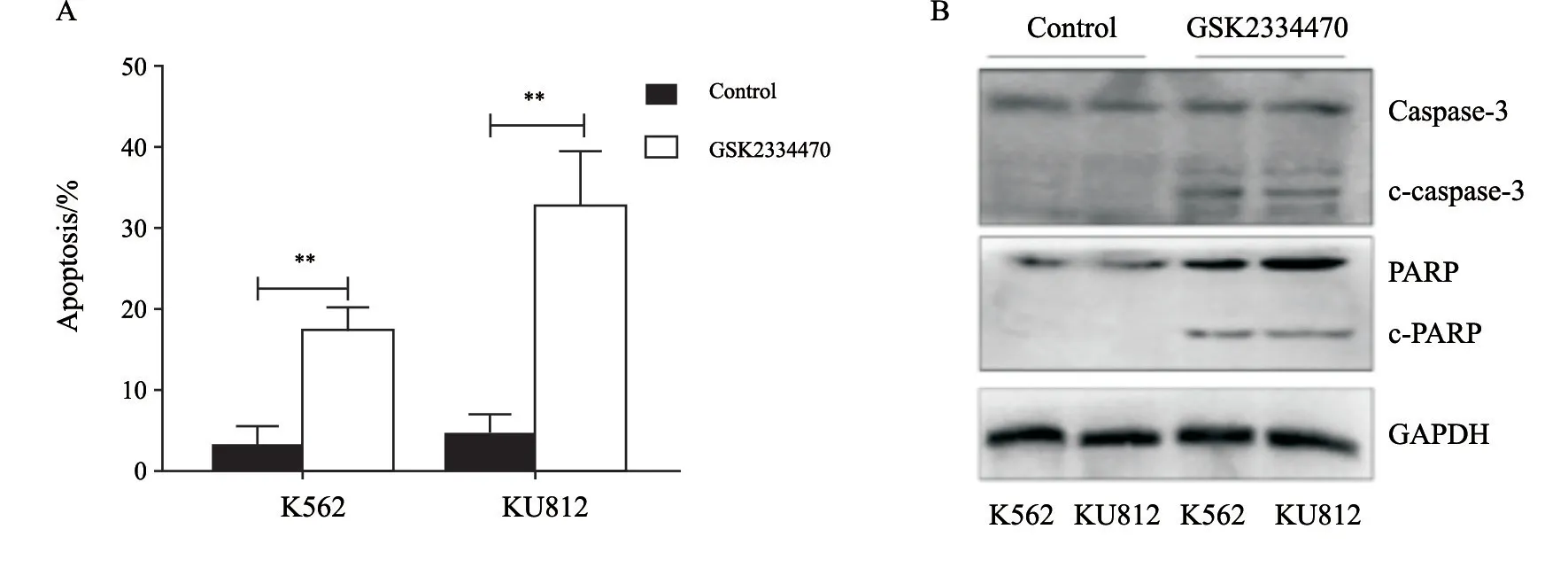
图3 流式细胞术检测GSK2334470对细胞的凋亡诱导作用(A);Western blot检测caspase-3及底物PARP的活化(B)Fig.3 Induction of apoptosis by GSK2334470 was detected by flow cytometry (A),and activation of caspase-3 and PARP were detected by Western blot (B)
2.4 抑制PDK1导致AKT活化降低
GSK2334470不能降低总PDK1表达水平,而是抑制其磷酸化,在K562细胞中,处理组PDK1磷酸化约降至对照组的39.4%(P<0.05);在KU812细胞中则更为明显,不足对照组的22.6%(P<0.01,图4A)。PDK1能够调节AKT磷酸化,因此,PDK1抑制后AKT磷酸化水平在两株细胞中也明显降低,在K562和KU812细胞中,分别降至对照组的31.4%(P<0.01)和42.6%(P<0.05)。AKT参与了多个下游分子的磷酸化调节,其中ASK1和GSK-3β的磷酸化水平均有明显下调。磷酸化ASK1含量在两株细胞中分别下降了约73.4%和86.9%(P<0.001),GSK-3β则分别下降了66.1%(P<0.01)和78%(P<0.001)。而作为GSK-3β下游的主要效应分子,总β-catenin及磷酸化β-catenin均没有明显变化,c-Myc作为GSK-3β/β-catenin通路的靶基因,其表达也未见明显变化(图4B)。
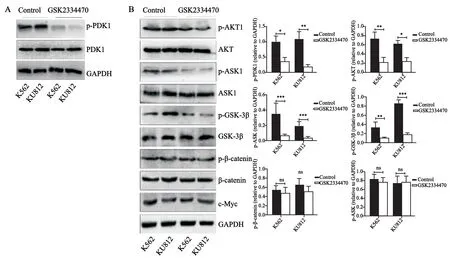
图4 Western blot检测GSK2334470处理后磷酸化及总PDK1变化(A);Western blot检测GSK2334470处理后ASK1磷酸化水平及AKTGSK-3β-β-catenin通路活化(B)Fig.4 Reduction in p-PKD1 and total PDK1 level was detected by Western blot (A),and activation of AKT-GSK-3β-β-catenin pathway was determined by Western blot (B)
2.5 ASK1/JNK/Bim介导细胞凋亡
磷酸化ASK1(Ser83)水平降低可促进其活性,因此检测ASK1下游JNK的磷酸化水平。两株细胞中磷酸化JNK在抑制剂处理后均有明显上调(K562:P<0.01,KU812:P<0.05),同时,作为JNK下游的Bcl-2家族促凋亡分子,Bim的磷酸化水平亦明显增加(K562:P<0.001,KU812:P<0.05)。另外,在K562细胞中,总Bim水平也明显上升(P<0.05),而在KU812中则不明显(图5)。
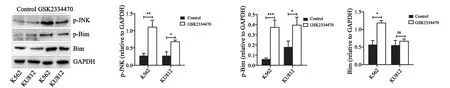
图5 Western blot检测GSK2334470刺激后磷酸化JNK、磷酸化Bim及总Bim水平Fig.5 Increase in phosphorylated JNK,phosphorylated Bim and total Bim under GSK2334470 stimuli was detected by Western blot
2.6 抑制PDK1增加imatinib敏感性
Imatinib单独作用对两株细胞的凋亡诱导率为20%~30%(K562为20.0%±3.95%),而与GSK2334470联合作用时,凋亡率均提高了1倍以上:K562细胞凋亡率为52.3%±6.77%(P<0.01);KU 812 为67.03%±8.94%(P<0.01)。GSK2334470能够显著提高细胞对imatinib的敏感性(图6)。
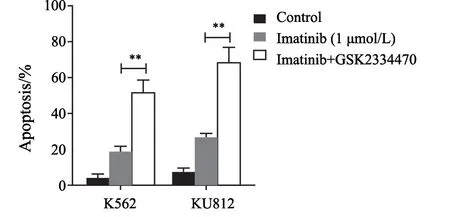
图6 GSK2334470提高细胞对imatinib的敏感性Fig.6 GSK2334470 promotes the sensitivity of cells to imatinib
3 讨 论
作为丝/苏氨酸蛋白激酶,PDK1是PI3K/AKT信号通路活化的重要激酶,可磷酸化激活AKT的Thr308位点[8]。后者活化后调节其下游多条信号转导通路,参与恶性肿瘤细胞增殖、侵袭等多种生物学行为。研究显示,PDK1在乳腺癌、结肠癌、急性白血病等多种肿瘤细胞中高表达,而抑制PDK1可有效地抑制肿瘤细胞增殖、侵袭[9-10]。因此,PDK1已经成为肿瘤治疗的新靶点。
本研究证实,PKD1在CML外周血单个核细胞中高表达。随后在两株CML细胞株中观察到,抑制PDK1具有明显的抑制细胞增殖和诱导细胞凋亡作用。抑制PDK1诱导的凋亡依赖于caspase通路。为进一步探讨抑制PDK1诱导细胞凋亡的分子机制,对AKT及ASK1信号两条通路进行活性分析。结果显示,PDK1失活后AKT及下游GSK-3β磷酸化水平明显降低,而作为GSK-3β下游的β-catenin的磷酸化水平却未见明显变化。同时作为AKT-GSK-3β通路的靶基因,c-Myc表达也未受影响。因此,推测有其他通路负责抑制PDK1引起的细胞凋亡行为。抑制PDK1后,ASK1磷酸化水平明显下调,提示其活性增加,这也被其下游JNK磷酸化水平上调所进一步证实。JNK可通过磷酸化激活Bim诱导细胞凋亡[11]。本研究结果显示,总Bim和磷酸化Bim水平均明显上调,故推测ASK1-JNK-Bim是负责细胞凋亡的主要效应分子。最后也证实抑制PDK1能够明显提高两株CML细胞对imatinib的敏感性,推测其可能具有逆转imatinib耐药的作用,然而因为缺乏耐药的细胞,未能更进一步证实。
综上所述,PDK1在CML细胞中的高表达参与了细胞逃避凋亡的生物学行为,有望成为CML治疗及克服耐药的新靶点。
