2,3-丁二酮介导的CF3SO2Na与烯烃的氧化三氟甲基化反应
2020-07-13徐文艺冯乙巳
徐文艺, 冯乙巳,2
(1. 合肥工业大学化学与化工学院; 2. 安徽省杂环实验室, 合肥 230009)
近几十年来, 约20%的药物分子和35%的农药中都含有氟原子[1~3], 不仅显著提高了有机药物的稳定性, 增加了化合物的溶解性, 而且也降低了生物的耐药性[4,5]. 在药物合成中, 为了利用氟原子的独特活性因子, 常将含氟基团引入有机物结构中以获得具有独特生物活性的化合物[6~8]. 三氟甲基(CF3)具有强吸电子性、亲脂性、代谢和化学稳定性, 因此, 以该基团作为氟源的药物相对于其它含氟化合物在结构改良上更具优势, 有更强的有机药物选择性[1], 广泛应用于医药、农药和新型功能材料等领域[9,10].
α-三氟甲基羰基化合物是化学合成和药物研究过程中重要的含氟中间体和产物[11], 传统上多采用酮或烯醇式化合物与自由基或亲电三氟甲基源反应的方法合成[12~15]. Mikami等[16]使用过量的nBuLi,iPr2OH和Ti(OiPr)4实现了钛酸盐烯醇式化合物的三氟甲基化反应. 在RCF3酮的钛烯醇化对脱氟稳定的基础上, Itoh等[17]将钛酸酯用于自由基三氟甲基化合成RCF3酮. 2013年, Hu等[18]采用Cu催化α-重氮酯与三氟甲基三氟硅烷(TMSCF3)的三氟甲基化反应, 在此体系中加入H2O促进α-重氮酯与碘进行离子交换, 形成少量的活性中间体. 但这些方法通常需要两步或多步反应完成, 反应条件比较苛刻, 低温条件有限, 且该类型反应使用的含酮类结构的烯醇类底物不易合成, 区域选择性较差. 因此, 近年来烯烃的直接氧化三氟甲基化法成为制备α-三氟甲基羰基衍生物的研究热点. 目前, Guo等[19]、Akita等[20]和Singh[21]等分别报道了Ru, Ir和MnO2催化的烯烃的酮三氟甲基化反应. Grushin等[22]首次开发了α-卤代酮的亲核三氟甲基化反应. Wu等[23]和Zhao等[24]报道了只在氧化体系下进行的三氟甲基化反应. Deb等[25]报道了在室温条件下用AgNO3和K2S2O8组成体系, 催化烯烃与CF3SO2Na反应得到α-三氟甲基苯乙酮. Itoh等[26]提出紫外光照射下2-氯蒽醌(2-Cl-AQN)催化的烯烃的氧化三氟甲基化反应. 尽管Ir和Ru等过渡金属催化剂在可见光中有较好的催化效率, 但这些金属配合物价格昂贵、制备复杂, 且具有潜在的毒性, 在大规模使用时会对环境和人体造成伤害. 因此, 急需探寻出一种替代金属催化剂的绿色、经济、简洁催化烯烃的酮三氟甲基化方法.
含有酮类结构(如丙酮和2,3-丁二酮等)的简单化合物是有机合成中常用的助溶剂, 也是最早出现的有机显色剂之一[27]. Li等[28]发现羰基氧的孤对电子从氧跃迁到羰基碳上后, 产生的缺电子羰基氧原子可以从CF3SO2Na中得到1个电子, 从而被还原, 同时得到三氟甲基自由基. 由于丙酮的吸收带处于330 nm以下的紫外波段, 而紫外线对人的眼睛和皮肤伤害很大, 故Li等[25]又进一步采用2,3-丁二酮作为自由基引发剂将波长范围延伸至可见光区域, 获得了到很好的催化效果. 基于此, 本文对2,3-丁二酮催化烯烃的氧化三氟甲基化反应进行了研究, 以苯乙烯和CF3SO2Na为原料合成了3,3,3-三氟-1-苯基丙-1-酮类化合物, 其结构经核磁共振波谱(NMR)等表征.
1 实验部分
1.1 试剂与仪器
苯乙烯(纯度99%)、三氟甲基亚磺酸钠(CF3SO2Na, 纯度99%)、2,3-丁二酮(2,3-Butanedione, 纯度≥99.0%)、丙酮(Acetone, 纯度99%)、四氢呋喃(THF, 纯度99.0%)、二氯甲烷(DCM, 纯度99.5%)、石油醚(PE, 纯度95.0%)、罗丹明B(Rhodamine B, 纯度95%)、曙红Y(Eosin Y, 纯度96.2%)和孟加拉玫瑰红(Rose bengal, 纯度90%)均购自阿拉丁化学试剂有限公司; 过氧化氢(H2O2, 分析纯)、过氧化氢叔丁醇(TBHP, 纯度70% in H2O)、荧光素(Fluorescein, 纯度90%)和甲基高氯酸盐(Mes-Acr-Me+ClO4, 纯度92%)均购自上海麦克林生化科技有限公司; 乙腈(MeCN, 纯度99.9%)、N,N-二甲基甲酰胺(DMF, 纯度99.9%)和1,4-二氧六环(1,4-Dioxane, 纯度≥99.9%)均购自安耐吉化学试剂有限公司; 无水硫酸钠(Na2SO4, 纯度99%)购自广东翁江化学试剂有限公司; 乙酸乙酯(EtOAc, 纯度99.9%)购自上海泰坦科技股份有限公司; 三联吡啶氯化钌六水合物[Ru(bpy)3Cl2, 纯度98%]购自上海易恩化学技术有限公司.
VNMRS600型超导核磁共振波谱(NMR)仪, 以CDCl3为溶剂, TMS为内标, 美国VARIAN公司; Trace 1300ISQ型气相色谱-质谱联用(GC-MS)仪, 苏州冷杉精密仪器有限公司; HSGF254型薄层层析硅胶板, 烟台江友硅胶开发有限公司; YRT-3型熔点仪, 天津天光光学仪器有限公司.
1.2 实验过程
目标化合物的合成路线如Scheme 1所示.
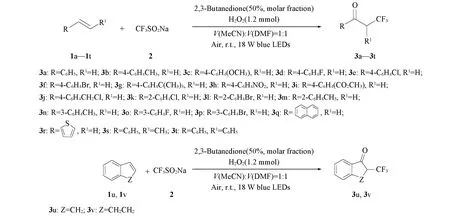
Scheme 1 Synthesis route of α-CF3-substituted ketones from styrenes
参照文献[21,23]方法合成目标化合物3a~3v. 向15 mL反应管中依次加入烯烃(0.2 mmol)、CF3SO2Na(0.5 mmol)、2,3-丁二酮(50%)、H2O2(1.2 mmol)和MeCN/DMF(体积比为1∶1)混合溶剂, 置于18 W蓝灯照射下反应18 h. 利用柱层析法对产物进行分离纯化, 洗脱剂为石油醚/乙酸乙酯(体积比100∶1), 得到目标化合物. 其理化数据和核磁波谱数据分别列于表1和表2.
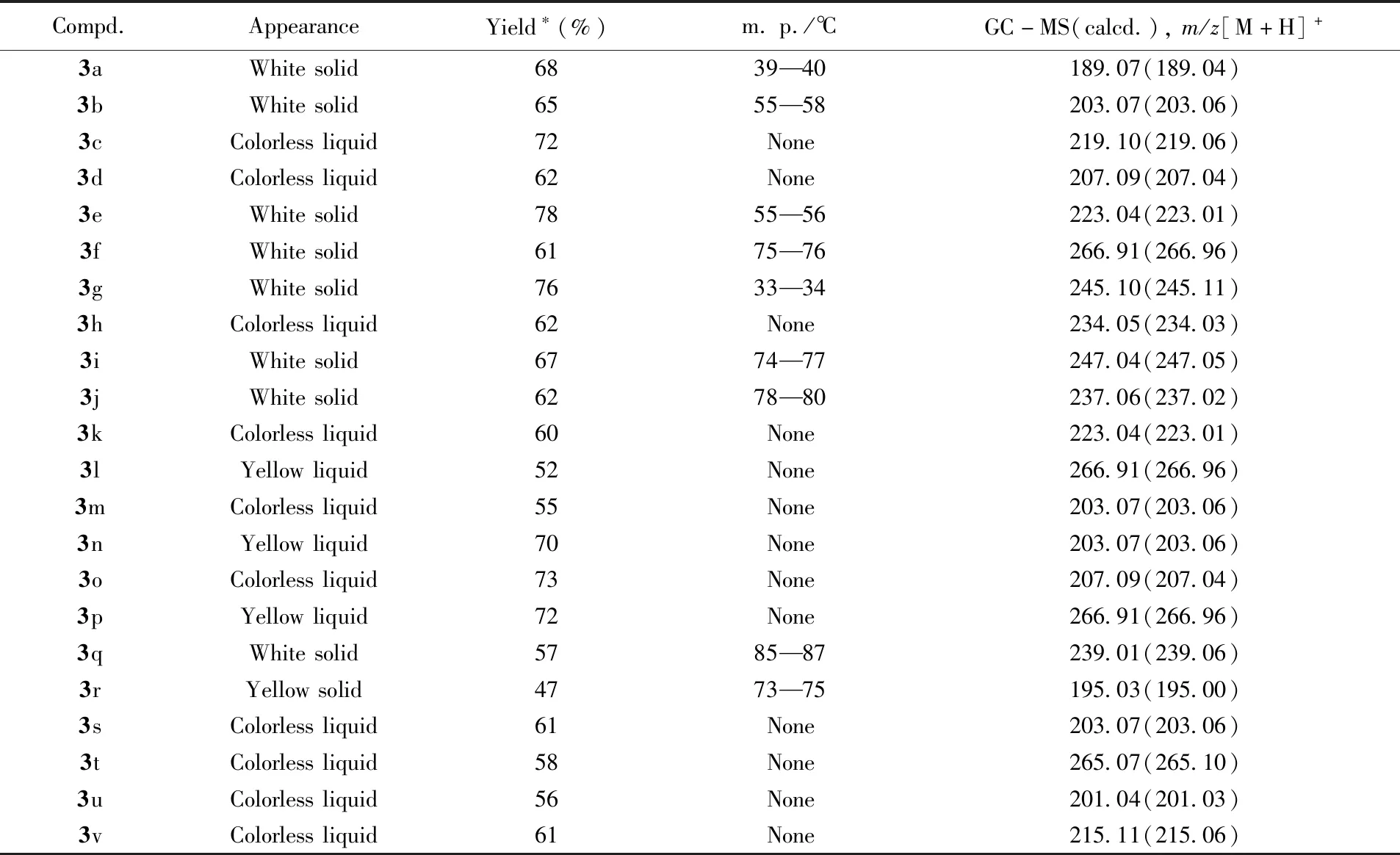
Table 1 Appearance, yields, melting points and GC-MS data for compounds 3a—3v
* Yield after purification by column chromatography on silica gel.
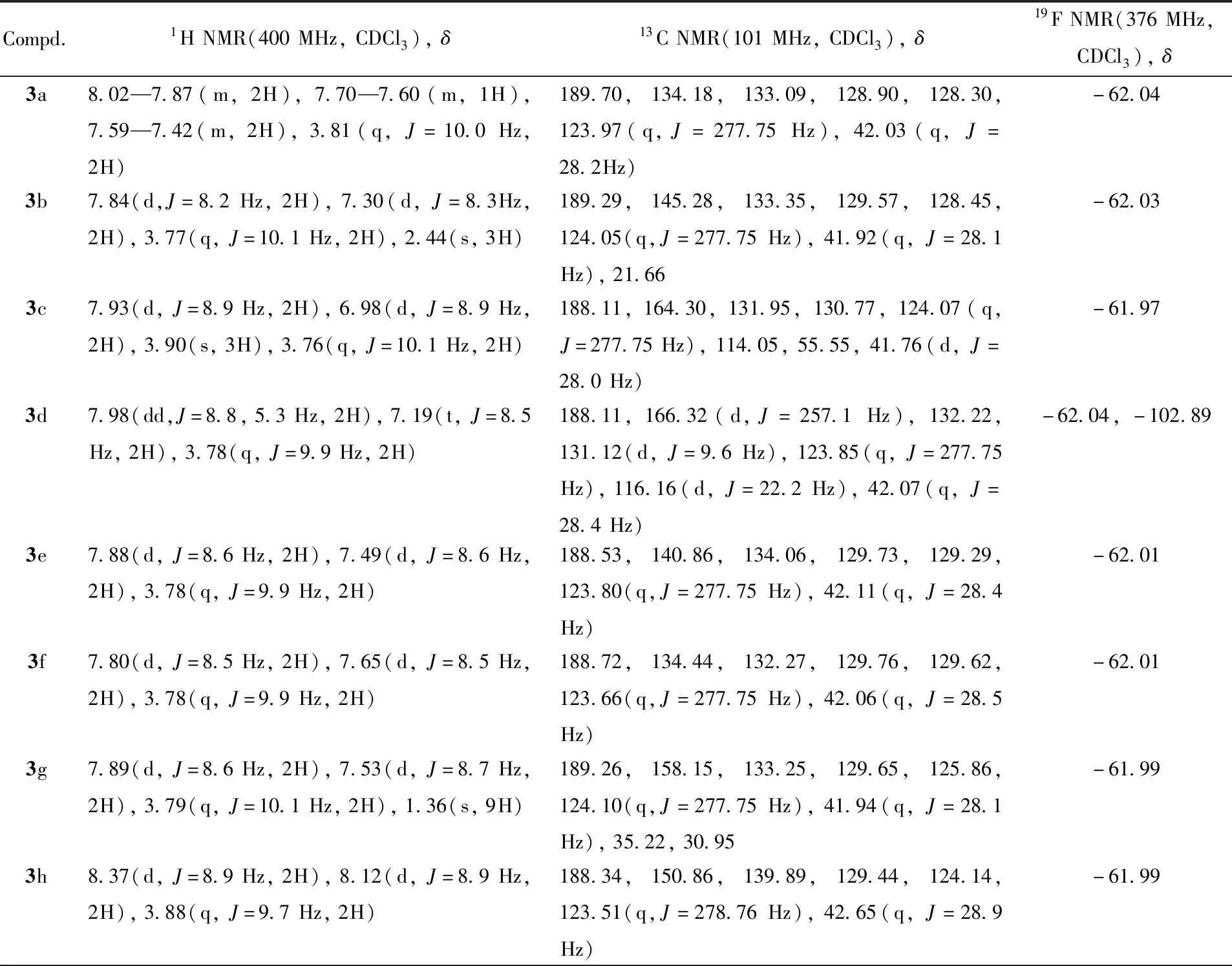
Table 2 1H NMR, 13C NMR and 19F NMR data for compounds 3a—3v
Continued
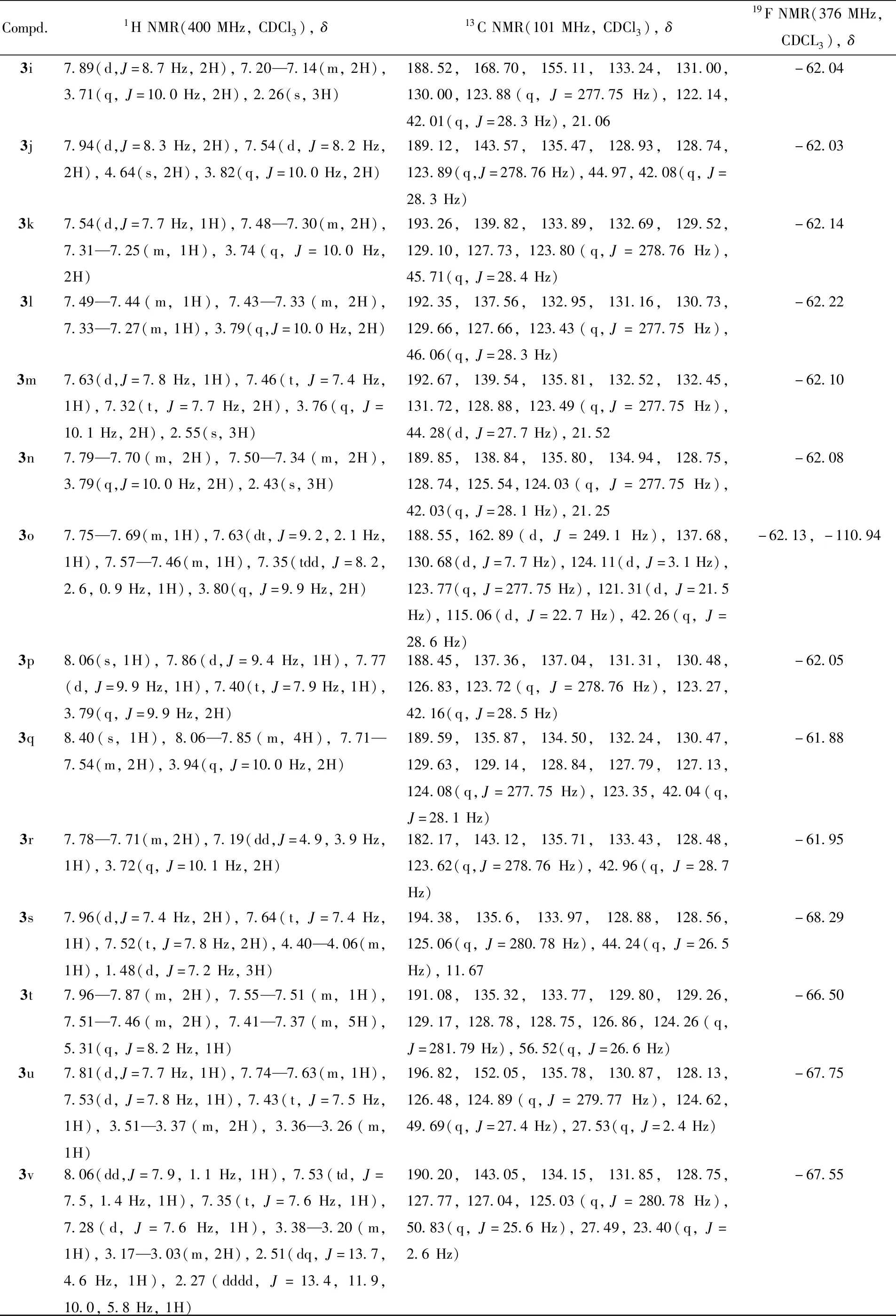
Compd.1HNMR(400MHz,CDCl3),δ13CNMR(101MHz,CDCl3),δ19FNMR(376MHz,CDCL3),δ3i7.89(d,J=8.7Hz,2H),7.20—7.14(m,2H),3.71(q,J=10.0Hz,2H),2.26(s,3H)188.52,168.70,155.11,133.24,131.00,130.00,123.88(q,J=277.75Hz),122.14,42.01(q,J=28.3Hz),21.06-62.043j7.94(d,J=8.3Hz,2H),7.54(d,J=8.2Hz,2H),4.64(s,2H),3.82(q,J=10.0Hz,2H)189.12,143.57,135.47,128.93,128.74,123.89(q,J=278.76Hz),44.97,42.08(q,J=28.3Hz)-62.033k7.54(d,J=7.7Hz,1H),7.48—7.30(m,2H),7.31—7.25(m,1H),3.74(q,J=10.0Hz,2H)193.26,139.82,133.89,132.69,129.52,129.10,127.73,123.80(q,J=278.76Hz),45.71(q,J=28.4Hz)-62.143l7.49—7.44(m,1H),7.43—7.33(m,2H),7.33—7.27(m,1H),3.79(q,J=10.0Hz,2H)192.35,137.56,132.95,131.16,130.73,129.66,127.66,123.43(q,J=277.75Hz),46.06(q,J=28.3Hz)-62.223m7.63(d,J=7.8Hz,1H),7.46(t,J=7.4Hz,1H),7.32(t,J=7.7Hz,2H),3.76(q,J=10.1Hz,2H),2.55(s,3H)192.67,139.54,135.81,132.52,132.45,131.72,128.88,123.49(q,J=277.75Hz),44.28(d,J=27.7Hz),21.52-62.103n7.79—7.70(m,2H),7.50—7.34(m,2H),3.79(q,J=10.0Hz,2H),2.43(s,3H)189.85,138.84,135.80,134.94,128.75,128.74,125.54,124.03(q,J=277.75Hz),42.03(q,J=28.1Hz),21.25-62.083o7.75—7.69(m,1H),7.63(dt,J=9.2,2.1Hz,1H),7.57—7.46(m,1H),7.35(tdd,J=8.2,2.6,0.9Hz,1H),3.80(q,J=9.9Hz,2H)188.55,162.89(d,J=249.1Hz),137.68,130.68(d,J=7.7Hz),124.11(d,J=3.1Hz),123.77(q,J=277.75Hz),121.31(d,J=21.5Hz),115.06(d,J=22.7Hz),42.26(q,J=28.6Hz)-62.13,-110.943p8.06(s,1H),7.86(d,J=9.4Hz,1H),7.77(d,J=9.9Hz,1H),7.40(t,J=7.9Hz,1H),3.79(q,J=9.9Hz,2H)188.45,137.36,137.04,131.31,130.48,126.83,123.72(q,J=278.76Hz),123.27,42.16(q,J=28.5Hz)-62.053q8.40(s,1H),8.06—7.85(m,4H),7.71—7.54(m,2H),3.94(q,J=10.0Hz,2H)189.59,135.87,134.50,132.24,130.47,129.63,129.14,128.84,127.79,127.13,124.08(q,J=277.75Hz),123.35,42.04(q,J=28.1Hz)-61.883r7.78—7.71(m,2H),7.19(dd,J=4.9,3.9Hz,1H),3.72(q,J=10.1Hz,2H)182.17,143.12,135.71,133.43,128.48,123.62(q,J=278.76Hz),42.96(q,J=28.7Hz)-61.953s7.96(d,J=7.4Hz,2H),7.64(t,J=7.4Hz,1H),7.52(t,J=7.8Hz,2H),4.40—4.06(m,1H),1.48(d,J=7.2Hz,3H)194.38,135.6,133.97,128.88,128.56,125.06(q,J=280.78Hz),44.24(q,J=26.5Hz),11.67-68.293t7.96—7.87(m,2H),7.55—7.51(m,1H),7.51—7.46(m,2H),7.41—7.37(m,5H),5.31(q,J=8.2Hz,1H)191.08,135.32,133.77,129.80,129.26,129.17,128.78,128.75,126.86,124.26(q,J=281.79Hz),56.52(q,J=26.6Hz)-66.503u7.81(d,J=7.7Hz,1H),7.74—7.63(m,1H),7.53(d,J=7.8Hz,1H),7.43(t,J=7.5Hz,1H),3.51—3.37(m,2H),3.36—3.26(m,1H)196.82,152.05,135.78,130.87,128.13,126.48,124.89(q,J=279.77Hz),124.62,49.69(q,J=27.4Hz),27.53(q,J=2.4Hz)-67.753v8.06(dd,J=7.9,1.1Hz,1H),7.53(td,J=7.5,1.4Hz,1H),7.35(t,J=7.6Hz,1H),7.28(d,J=7.6Hz,1H),3.38—3.20(m,1H),3.17—3.03(m,2H),2.51(dq,J=13.7,4.6Hz,1H),2.27(dddd,J=13.4,11.9,10.0,5.8Hz,1H)190.20,143.05,134.15,131.85,128.75,127.77,127.04,125.03(q,J=280.78Hz),50.83(q,J=25.6Hz),27.49,23.40(q,J=2.6Hz)-67.55
2 结果与讨论
2.1 反应条件优化
以苯乙烯(1a)与三氟甲基亚磺酸钠(2a)为模型反应底物, 考察了不同催化剂、氧化剂、溶剂及时间等因素对反应产率的影响. 反应条件优化结果见表3.
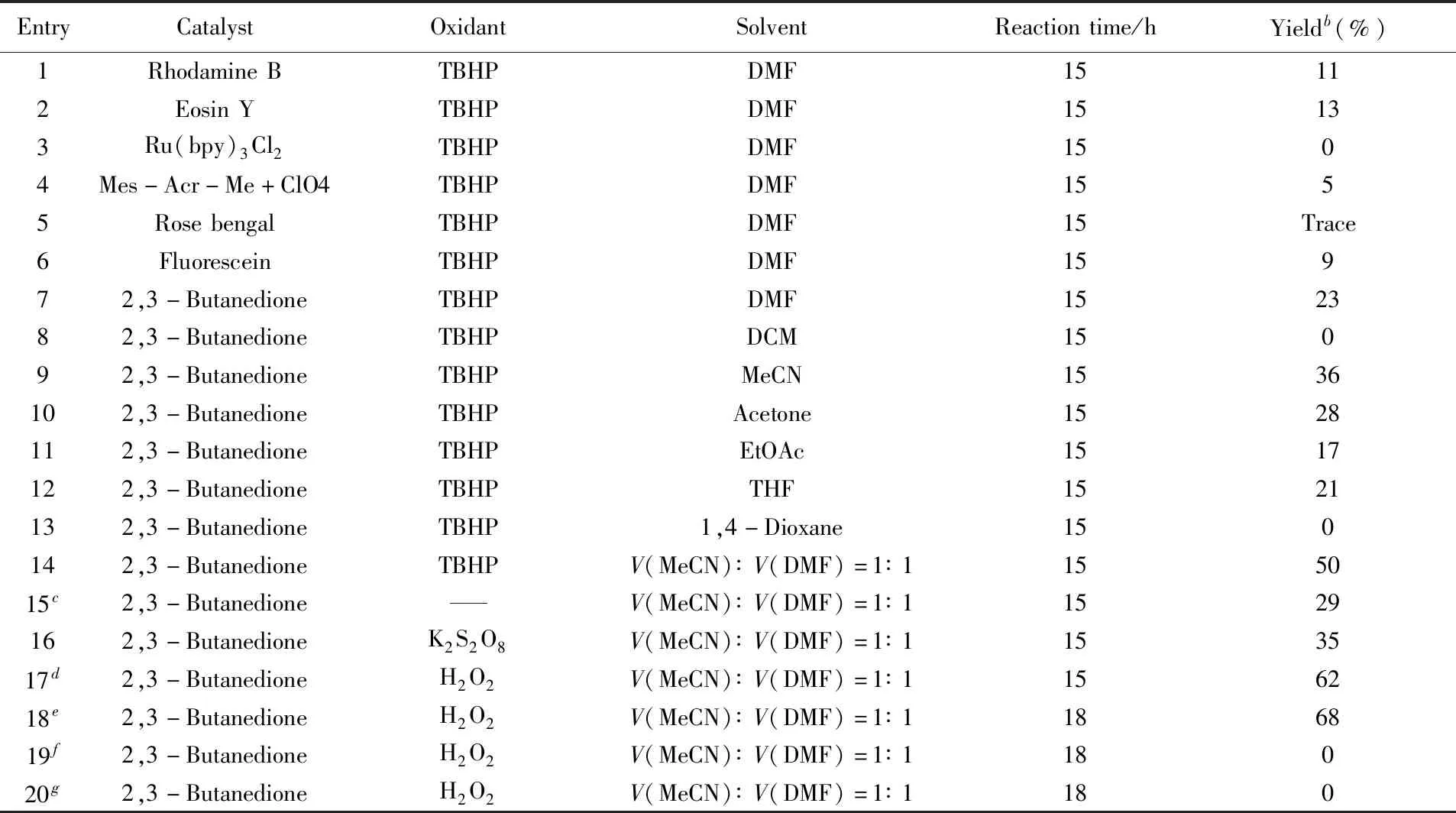
Table 3 Conditions optimization of model reactiona
a. Reaction conditions: styrene(1a, 0.2 mmol), CF3SO2Na(2a, 0.5 mmol), 2,3-butanedione(50%, molar fractopm), oxidant(1.2 mmol), MeCN/DMF solution(volume ratio 1∶1, 2 mL) at room temperature under blue LED(18 W) in air for 18 h;b. isolated yield;c. O2atmosphere;d. six equivalents of oxidant was added;e. reaction performed for 18 h;f. under white LED;g. in dark.
在其它条件不变的情况下, 首先对不同有机光催化剂进行了筛选. 结果表明, 与玫瑰红、荧光素和Eosin Y等常用催化剂相比, 2,3-丁二酮表现出较高的催化活性, 并以23%的收率获得目标产物3a(表3中Entry 7); 而以Ru(bpy)3Cl2为光催化剂时, 仅得到痕量的目标产物3a(表3中Entry 3). 对反应溶剂进行了筛选优化, 以V(MeCN)∶V(DMF)=1∶1混合溶剂反应时, 产物3a的收率显著提高至50%(表3中Entry 14). 与混合溶剂相比, 单一溶剂对反应效果的提升并不理想(表3中Entries 9~12), 尤其是以DCM和1,4-Dioxane作溶剂时, 无目标产物生成(表3中Entries 8和13). 其次, 筛选了几种常见的氧化剂. 以TBHP作为氧化剂时, 最高产率为50%(表3中Entry 14). 当以K2S2O8为氧化剂或在氧气条件下, 化合物3a的产率均有所降低(表3中Entries 15和16). 实验中发现, 当使用H2O2为氧化剂并将其用量提高到1.2 mmol时, 化合物3a的产率可提高至62%(表3中Entry 17). 以上实验结果证明, H2O2为该反应的最佳氧化剂. 另外, 将反应时间从最初的15 h延长至18 h后, 收率略微提升至68%(表3中Entry 18), 继续延长反应时间产率变化不大, 据此推断适当地延长反应时间对该反应产率的提高有推动作用. 对反应所需光源进行了筛选, 在白光或者黑暗条件下均检测不到目标产物的生成(表3中Entries 19和20), 说明蓝光能使2,3-丁二酮变成激发态, 与CF3SO2Na发生电子转移产生三氟甲基自由基. 最后, 对化合物1a与2a的摩尔比进行了优化, 发现n(1a)∶n(2a)=2∶5时反应效果最好. 通过对模型反应各种影响因素的优化和筛选, 确定了反应的最佳条件: 苯乙烯(0.2 mmol), CF3SO2Na(0.5 mmol), 2,3-丁二酮(摩尔分数50%), H2O2(1.2 mmol), 溶剂为乙腈/水(V∶V=1∶1, 2 mL), 18 W蓝灯作为光源, 于空气中室温下反应18 h, 收率达到68%.
2.2 底物适应性
根据表3所得最优模型的反应条件, 对反应底物的适用范围进行了研究, 结果见表1. 首先, 考察了苯环邻、间、对位上的取代基对反应的影响. 结果表明, 在苯环的对位上具有供电子基团(如Me, MeO,t-Bu和MeO2C)的芳基烯烃与CF3SO2Na反应时, 分别以良好收率获得产物3b(65%),3c(72%),3g(76%)和3i(67%); 对位为吸电子基团如三卤元素的产物3d,3e和3f及硝基的产物3h等, 产率为61%~78%. 由此可见, 无论是含有供电子基团还是吸电子基团的芳基烯烃与CF3SO2Na反应时, 都具有良好的活性. 还考察了苯环上邻位和间位取代基的产物的反应性, 发现间氟(3o)、间溴(3p)、间甲基(3n)苯乙烯的酮三氟甲基化产物产率均在70%以上, 而邻位取代基(化合物3k,3l和3m)的反应性较差, 最高产率也只有60%, 这是由于邻位取代基产物的产率受空间位阻效应的影响. 此外, 萘乙烯的酮三氟甲基化产物(3q)的产率也仅有57%.
为了进一步拓宽底物范围, 还对其它烯烃类底物进行了探索, 包括杂环类和环烯烃类. 研究发现, 当使用2-乙烯基噻吩为原料时, 仅以47%的产率获得产物3r. 需要指出的是,α,β-取代的烯烃产物在合成化学中普遍存在, 因此, 选择(E)-β-甲基苯乙烯和(E)-1,2二苯乙烯与CF3SO2Na进行反应, 分别以61%和58%的收率获得产物3s和3t. 在此优化条件下, 还探究了环烯烃的反应性, 以中等收率将1,2-二氢茚和1,2-二氢萘转化为含CF3的环状酮3u(56%)和3v(61%).
2.3 反应机理
结合模型反应优化结果和文献[21,24]报道结果, 推测该反应可能涉及CF3自由基机理, 因而设计了自由基捕捉实验进行验证. 在优化条件下, 加入0.5 mmol 2,2,6,6-四甲基哌啶氮氧化物(TEMPO)自由基捕捉剂, 进行模型反应研究(见Scheme 2).

Scheme 2 Synthesis route ofTEMPO radical capture experiment
加入TEMPO捕捉剂后, GC-MS表征结果显示未检测到产物3a的离子峰, 但检测到TEMPO-CF3自由基捕捉剂产物, 说明该反应机理为自由基反应.
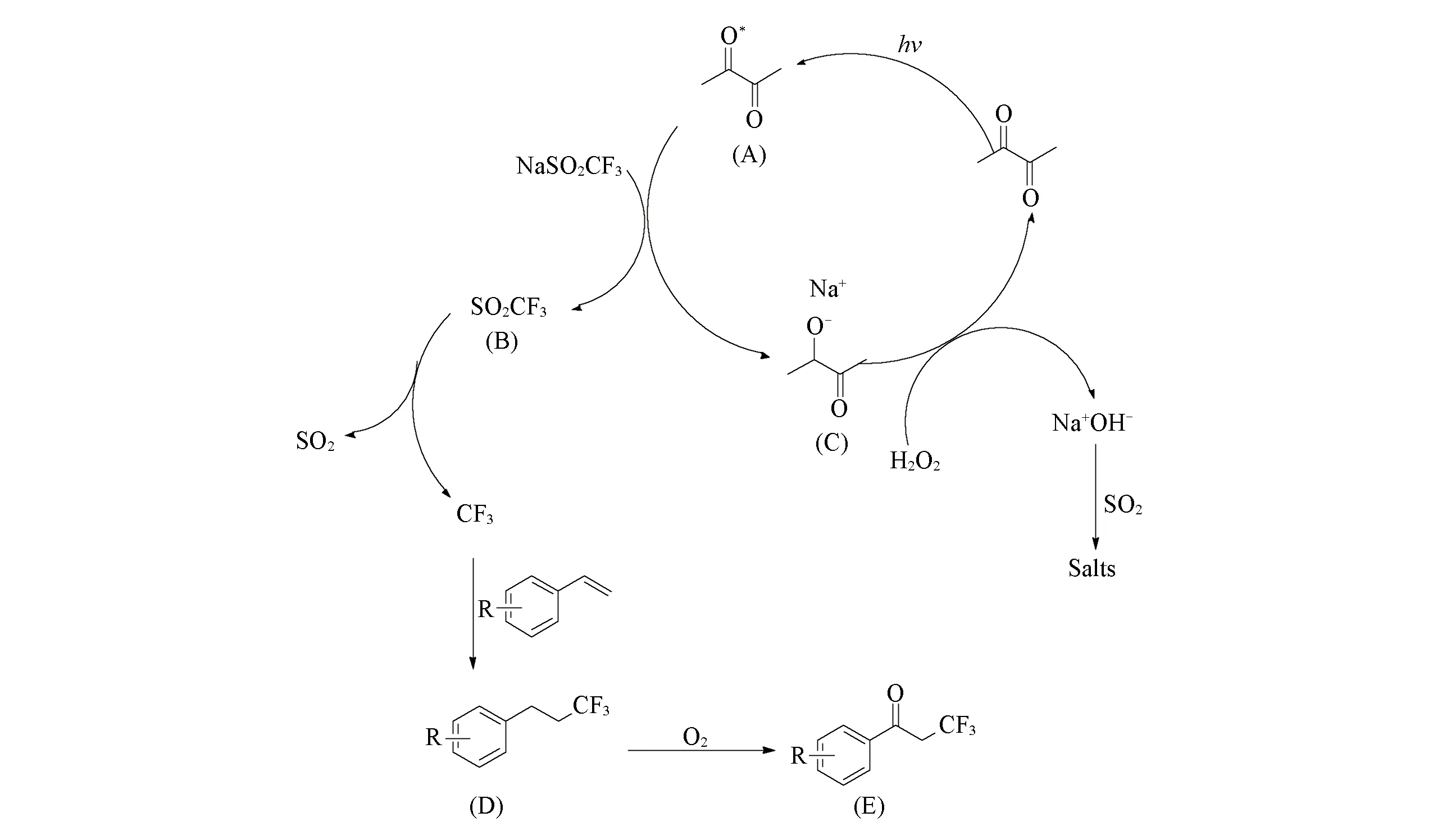
Scheme 3 Possible mechanism of oxidative trifluoromethylation of CF3SO2Na with olefins catalyzed by diacetyl
基于文献[22,25]报道及自由基捕捉实验结果, 提出了可能的反应机理(见Scheme 3). 首先, 可见光激发2,3-丁二酮发生n,π*轨道跃迁变为激发态的A, 随后A与CF3SO2Na反应, 夺取三氟甲磺酸负离子的1个电子生成三氟甲磺酸自由基B, 继而脱除二氧化硫生成三氟甲基自由基, 同时A转化成中间体C, 然后C被过氧化氢氧化成2,3-丁二酮, 参与催化循环; 过氧化氢转化为氢氧根离子, 结合钠离子成为氢氧化钠, 氢氧化钠与脱除的二氧化硫反应生成盐; 生成的三氟甲基自由基与苯乙烯类化合物反应得到苄基自由基D, 再被O2进一步氧化成酮, 从而得到目标产物E.
3 结 论
采用2,3-丁二酮为光催化剂, 过氧化氢为氧化剂, MeCN/DMF(体积比1∶1)为反应溶剂, 以各种苯乙烯类化合物、环状烯烃以及1,1-二取代烯烃为原料, 在18 W蓝灯照射下与三氟甲基磺酸钠进行氧化三氟甲基化反应, 并以中等至良好的收率合成了23种具有区位高选择性的α-三氟甲基酮类化合物. 机理研究表明, 该反应涉及三氟甲基自由基参与, 并在过氧化氢的协同下进行催化剂的循环利用以及苄基自由基的氧化. 与其它昂贵金属催化剂或危险的过氧化物相比, 利用丁二酮催化CF3SO2Na为氟源的可见光诱导三氟甲基化反应, 具有反应条件温和、底物适用范围广及催化特性优良等优点, 为经济、有效地大规模合成三氟甲基化化学品提供了一条更环保、高效的途径, 在医学以及工农业上具有一定的应用前景.
