团簇Co4P的成键及HOMO和LUMO轨道贡献
2020-07-13方志刚
许 友, 方志刚, 廖 薇, 秦 渝
(辽宁科技大学 化学工程学院, 辽宁 鞍山 114051)
0 引 言
非晶态合金内部原子的排列具有短程有序、 长程无序的特点, 微观上具有液体的无序原子结构, 宏观上又具有固体的刚性, 是一种亚稳态材料[1-2]. 因此, 非晶态合金具有其他合金所不具备的优良特性, 甚至在某些方面可以替代贵金属, 为工业生产降低成本. 其中, Co-P作为非晶态合金研究中的热点, 同样具有一系列优良特性. Liu Q等[3]验证了Co-P合金改性的ZnLn2S4复合材料在可见光照射下的光催化剂制氢活性比含有Pt(质量分数为1%)的ZnLn2S4复合材料高; Ryu J等[4]论述了CoP纳米粒子在碱性溶液中具有较高效率的析氢作用, 甚至可以与贵金属铱相媲美; Oh S等[5]在高阴极电流密度下制备Co-P泡沫型催化剂, 其过电位与Pt/C相当, 且在KOH碱性溶液中具有比Ir/C和RuO2更好的析氧(OER)活性. 由此可见, Co-P体系非晶态合金替代贵金属已可应用于实际生产中. 此外, Co-P体系的非晶态合金还具有成为优良镀层的潜能, Li R等[6]通过脉冲反向电流(PRC)在氯基镀液中制备出表面光滑、 具有无定形结构以及耐磨性和耐蚀性的钴磷镀层. 而且, 以可再生能源为原料的电催化生物质气化是生产可持续非化石碳产品的理想途径, Jiang N等[7]采用电沉积法制成的Co-P非晶态合金可作为5-羟甲基糠醛(HMF)以及2,5-呋喃甲酸(FDCA)的有效电催化剂, 这为能源短缺问题提供了一种新的解决办法. 此外, Co-P体系非晶态合金在医学方面也具有一定的研究价值, 如Jin L等[8]将Co-P体系非晶态合金用于癌症的治疗.
尽管Co-P体系的非晶态合金具有众多的优良性质, 但目前对其微观角度的理论研究甚少. 本文从微观角度对团簇Co4P各优化构型的成键以及HOMO和LUMO的轨道贡献率进行分析, 弥补了Co-P体系微观理论的知识空白, 并且为今后Co-P体系的研究提供了一定的理论依据.
1 理论和方法
以拓扑学原理[9]为理论依据, 设计出团簇Co4P所有可能存在的初始构型, 运用密度泛函理论(Density Functional Theory, DFT)[10-11]方法, 在B3LYP/Lanl2dz水平下对团簇Co4P的所有初始构型进行全参数优化和数据计算, 计算中对P加极化函数ξP.d=0.55[12], 并且采用与文献[13]相同的计算方法. 所有计算过程均在启天M4390计算机上用Gaussian09程序完成.
完成所有计算后再对不稳定的构型进行排除, 最终得到6种优化构型, 其中二、 四重态各为3种. 图 1 即为所得的6种优化构型图, 可知团簇Co4P具有两种稳定的空间结构, 分别为三角双锥(1(4), 2(2), 3(4))和戴帽三角锥(1(2), 2(4), 3(2)), 其中右上标代表自旋多重度. 能量最低的构型1(2)为0 kJ/mol, 其余构型均基于构型1(2)计算得出相对能量并按大小进行排序, 各优化构型的能量大小排序为: 1(2)<1(4)<2(2)<2(4)<3(2)<3(4), 具体数值见图 1.

图 1 团簇Co4P的6种优化构型Fig.1 6 optimized configurations of cluster Co4P
2 结果与讨论
2.1 键长分析
对键长进行分析可以进一步了解化学成键的强弱, 键长越短, 键能越大, 成键越稳定. 表 1 列出了各优化构型原子间的键长数据, 其中Co-P和Co-Co分别表示Co与P之间的平均键长和Co与Co之间的平均键长, 其范围分别为0.261~0.278 nm 和0.261~0.288 nm. Co-P键和Co-Co键的平均键长变化区间较小, 这表明各构型键长均处于相对稳定的状态. 当键长小于其所对应的原子半径之和时, 原子间易形成重叠部分从而有利于成键. 在团簇Co4P中, 由于Co与P的原子半径分别为0.125 nm和0.110 nm, 所以Co和P原子之间的半径和为0.235 nm, Co和Co原子之间的半径和为0.250 nm, 其值均小于对应键型的平均键长, 这在直观上可能产生各原子之间成键强度较弱的思想误区, 其实是由于数值整体平均化所导致的. 仔细分析表 1 中Co-P之间的键长后, 可以发现除构型1(2), 1(4), 2(4), 3(2)的Co3-P键和构型2(2)的Co4-P键以及构型3(4)的Co1-P键的键长大于Co和P原子半径之和外, 其余构型各Co原子与P原子之间所形成键(Co1-P, Co2-P, Co3-P, Co4-P)的键长均小于Co和P原子半径之和, 说明在整体上各Co原子与P原子之间是易于成键的. 对Co-Co之间的键长进行分析, 发现除构型2(2), 3(2)的Co2-Co4键和构型3(4)的Co1-Co2键的键长小于Co和Co原子半径和外, 其余构型各Co原子之间所形成的键(Co1-Co2, Co1-Co3, Co1-Co4, Co2-Co3, Co2-Co4, Co3-Co4)的键长均大于或等于Co和Co原子半径和, 这表明在整个团簇Co4P中各Co原子之间是较难成键的.
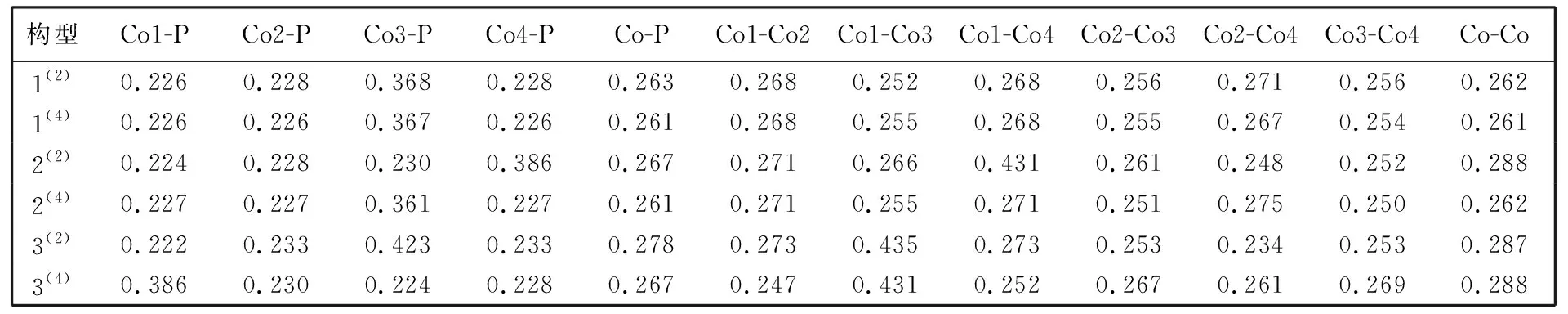
表 1 团簇Co4P各构型的键长
为了更直观地对各优化构型的键长进行分析, 作出图 2 所示的各优化构型键长平均值的折线图.

图 2 团簇Co4P各优化构型的平均键长Fig.2 Average bond length of each optimized configuration of cluster Co4P
由图 2 可知构型1(2), 1(4), 2(4)的Co-P键以及Co-Co键的平均键长均处于相对较低的水平, 可见这些构型在成键稳定上占据较高的优势; 构型3(2)的Co-P键和Co-Co键的平均键长则均处于相对较高的水平, 说明该构型总体上键长较长, 不利于成键, 成键相对不稳定; 虽然构型2(2)和3(4)的Co-P键的平均键长所处水平不高, 但其Co-Co键的平均键长大, 这对成键的稳定同样具有一定的不利影响.
2.2 键级分析
键级是分子轨道法中表示相邻两个原子成键强度的表达形式[14-15], 其值为正数时, 成键轨道上的电子称作成键电子, 它使团簇体系的能量降低, 且利于形成稳定的键; 其值为负数时, 所形成的反键轨道上的电子称作反键电子, 它使体系的能量升高, 不利于形成稳定的键. 可见, 键级是衡量化学键相对强弱的一个重要参数, 其正值越大, 成键越稳定, 若键级值为负则起反键作用, 不利于成键.
表 2 列出了各优化构型中不同键型的键级值, 其中Co-P和Co-Co分别表示Co与P之间的平均键级和Co与Co之间的平均键级, 其变化范围分别为0.199~0.205和-0.013~0.017. 可以发现Co-P键的键级平均值较大且其变化区间较小, 说明Co-P键有较强且稳定的成键作用; 而Co-Co键的平均键级较低, 不利于形成稳定的键, 因此其对构型的稳定性贡献较小. 此外, 部分构型(1(2), 1(4), 2(4))的Co-Co键级数值为负, 起反键作用, 更加不利于成键.
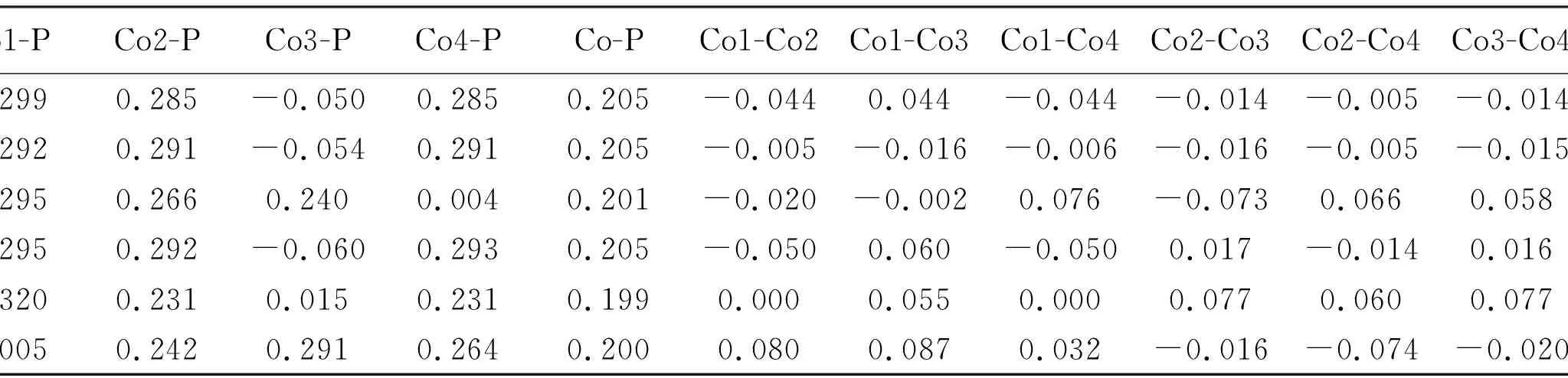
表 2 团簇Co4P各优化构型的各键键级
表 3 列出了团簇Co4P所有优化构型中各成键键级占总成键键级的百分比. 显然, Co-P键成键键级占总成键键级的比例(81.578%~100%)远大于Co-Co键 (0%~18.422%), 说明各优化构型中金属与非金属之间所形成的Co-P键对团簇Co4P的成键贡献较大. 而且, 在构型1(2), 1(4), 2(4)中Co-P成键键级占总成键键级的百分比达到了100%, 构型3(2)所占百分比最低, 但也达到了81.578%, 可见Co-P键对团簇Co4P的稳定性贡献较大. 对金属与金属之间所成的Co-Co键的键级进行分析发现, 除构型3(2)外, 其余构型的Co-Co键的键级占比均低于8%, 说明Co-Co键对团簇Co4P的成键稳定性贡献极小. 换言之, 团簇Co4P中Co-P键更容易成键, 而Co-Co键更不易成键, 其中在构型1(2), 1(4), 2(4)中Co-Co键最难形成. 综上所述, 在团簇Co4P中, 非金属与金属所形成的Co-P键在对团簇Co4P的成键贡献中占主导地位.

表 3 团簇Co4P优化构型中各键键级占总成键键级的百分比
2.3 态密度分析
态密度可以作为能带结构的一种可视化结果[16], 通过对态密度进行分析可以了解团簇Co4P的成键性质. 分波态密度和总态密度均是通过曲线的形式表达原子各轨道在一定能量范围内的电子分布情况, 前者针对的是单个原子, 后者则是对所有原子的综合分析. 图 3 为团簇Co4P各优化构型的分波态密度(PDOS)图与总态密度(TDOS)图, 其中竖直虚线表示Fermi能级, 各原子轨道对应曲线表示相应能量范围内的电子分布状态.
对图 3 中各构型的态密度图进行分析可知: 构型1(2)Fermi能级左侧峰A主要由Co-3d轨道和P-3p轨道贡献, 右侧峰B主要由Co-4p轨道、 Co-3d轨道和P-3p轨道贡献, 峰C主要由Co-4p轨道和P-3p轨道贡献; 构型1(4)Fermi能级左侧峰A主要由Co-3d轨道和P-3p轨道贡献, 右侧峰B主要由Co-4p轨道和P-3p轨道贡献; 构型2(2)Fermi能级左侧峰A主要由Co-3d轨道和P-3p轨道贡献, 右侧峰B主要由Co-4p轨道、 Co-3d轨道和P-3p轨道贡献, 峰C主要由Co-4p轨道和P-3p轨道贡献; 构型2(4)Fermi能级左侧峰A主要由Co-3d轨道和P-3p轨道贡献, 右侧峰B主要由Co-4p轨道和P-3p轨道贡献; 构型3(2)Fermi能级左侧峰A和峰B均主要由Co-3d轨道和P-3p轨道贡献, 右侧峰C主要由Co-4p轨道、 Co-3d轨道和P-3p轨道贡献; 构型3(4)Fermi能级左侧峰A主要由Co-3d轨道和P-3p轨道贡献, 右侧峰B主要由Co-4p轨道、 Co-3d轨道和P-3p轨道贡献, 峰C主要由Co-4p轨道和P-3p轨道贡献. 综合看来, 在团簇Co4P各优化构型的态密度图中, Fermi能级左侧峰A均由Co-3d轨道和P-3p轨道贡献, 且其峰值较高, 可见所有优化构型的成键方式中均存在p-d轨道的强杂化作用, 且构型3(2)Fermi能级左侧峰B同样由Co-3d轨道和P-3p轨道贡献, 说明构型3(2)内部成键过程中p-d杂化作用更为突出. 另外, 构型1(2), 3(4)Fermi能级右侧的峰B均由Co-4p轨道、 Co-3d轨道和P-3p轨道贡献, 可见这两种构型内部还存在一定的p-p-d杂化作用. 对各优化构型Fermi能级右侧的最高峰进行分析, 可以发现构型1(2), 1(4), 2(2), 2(4), 3(4)的Fermi能级右侧最高峰均由Co-4p轨道和P-3p轨道贡献, 可见这些构型内部存在着p-p杂化; 构型3(2)的右侧最高峰则由Co-4p轨道、 Co-3d轨道和P-3p轨道贡献, 说明该构型内部存在一定的p-p-d杂化作用.

图 3 团簇Co4P各优化构型的分波态密度图和总态密度图Fig.3 Diagrams of partial and total densities of states for various optimized configurations of cluster Co4P
综合分析各构型的分波态密度图, 可以发现Co-P键的形成方式有p-d, p-p, p-p-d 3种, 并且Co-3d轨道与P-3p轨道之间存在较大的重叠区间, 形成了较强p-d杂化作用, 有利于促进Co-P键的形成. 此外, Co-4s轨道、 P-3s轨道及P-3d轨道对团簇Co4P的的成键贡献极小.
2.4 HOMO和LUMO轨道的贡献
分子最高占据轨道HOMO以及分子最低未占据轨道LUMO决定了分子的电子得失和转移能力, 进而影响化学反应的程度. 表 4 列出了各优化构型中Co原子和P原子对HOMO和LUMO轨道的贡献率, 从而准确地预测出团簇Co4P的潜在催化活性位点.
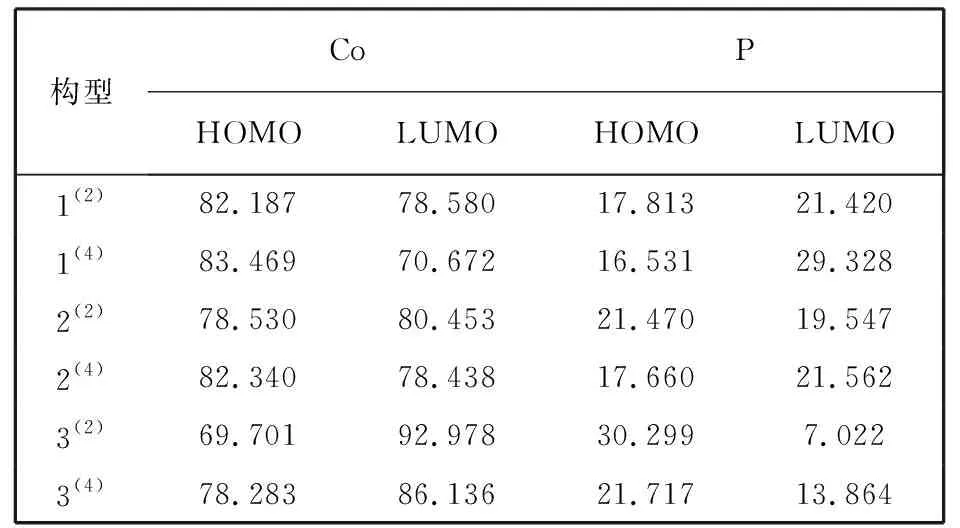
表 4 团簇Co4P中Co、 P原子对HOMO及LUMO轨道的贡献率
由表 4 可知, Co-HOMO轨道的贡献率范围为69.701%~83.469%, Co-LUMO轨道的贡献率范围为70.672%~92.978%, 其中构型1(2), 1(4), 2(4)中Co-HOMO轨道贡献率均超过82%, 构型2(2), 3(2), 3(4)中Co-LUMO轨道贡献率均高于80%; P-HOMO轨道贡献率范围为 16.531%~30.299%, P-LUMO轨道的贡献率范围为 7.022%~29.328%. 可见Co原子的HOMO和LUMO轨道贡献率均处于较高水平, 说明当团簇Co4P在化学反应中起催化作用时, 金属原子Co为主要贡献者, 该原子是潜在的催化活性位点.
为了更直观地体现团簇Co4P中各原子的HOMO和LUMO轨道贡献率的具体情况, 依据表 4 数据作出图 4 所示的折线图. 显然, Co原子的HOMO和LUMO轨道贡献率均高于P原子. 分析图 4 可以发现, Co-HOMO轨道与Co-LUMO轨道之间以及P-HOMO轨道与P-LUMO轨道之间均存在一定的拮抗作用; Co-HOMO轨道与P-LUMO轨道之间以及Co-LUMO轨道与P-HOMO轨道之间均存在一定的协同作用.

图 4 团簇Co4P中Co、 P原子的HOMO及LUMO轨道贡献率Fig.4 HOMO and LUMO orbitals contribution rates of Co and P atoms in cluster Co4P
3 结 论
本文从键长、 键级、 态密度图3个方面对团簇Co4P的成键进行分析, 并通过Co、 P原子的HOMO和LUMO轨道贡献率对团簇Co4P的催化活性位点进行探寻, 得到如下结论:
1) 键长分析: 通过将键长与相对应的原子间半径和进行对比, 得知Co-P键具有较强的成键作用, 对团簇Co4P构型的稳定性提供了较大贡献.
2) 键级分析: 对团簇Co4P的键级进行分析, 发现在成键贡献上Co-P键占主导地位, 极大地促进了团簇Co4P的稳定性.
3) 态密度分析: Co-4s轨道、 P-3s轨道及P-3d轨道对团簇Co4P的贡献极小; Co-3d轨道与P-3p轨道之间存在较大的重叠区间, 形成了较强p-d杂化作用, 促进Co-P键的形成; Co-P键的形成方式有p-d, p-p, p-p-d 3种.
4) HOMO及LUMO轨道贡献分析: 在团簇Co4P中, 金属原子Co为潜在的催化活性位点; Co的HOMO轨道与LUMO轨道之间以及P的HOMO轨道与LUMO轨道之间均存在一定的拮抗作用; Co的HOMO轨道与P的LUMO轨道之间以及Co的LUMO轨道与P的HOMO轨道之间均存在一定的协同作用.
