鲜鱼腥草UPLC特征图谱及5种指标成分含量测定研究
2020-07-08曾昭君邓李红黄梦婷刘燎原程学仁
徐 杰,曾昭君,邓李红,魏 梅,黄梦婷,刘燎原,程学仁
广东一方制药有限公司 广东省中药配方颗粒企业重点实验室,佛山 528244
鲜鱼腥草为三白草科植物蕺菜(HouttuyniacordataThunb.)的新鲜全草,性辛,味微寒,具有清热解毒,消痈排脓,利尿通淋的功效[1],是常见的“药膳兼用型”大宗中药材之一,具有广阔的经济和药用价值。研究表明,鱼腥草主要的活性成分为挥发油、有机酸和黄酮类化合物,其中绿原酸、芦丁、槲皮苷等是其质量控制的重要指标[2-4]。显然,单一成分定量分析难以全面反映鲜鱼腥草药材的质量。
中药特征图谱是中药整体性的化学表征,在中药质量评价方面应用广泛[5]。目前已有鲜鱼腥草HPLC特征图谱研究的报道[6],但该方法检测时间长达70~90 min,供试品制备方法复杂、分析耗时长、试剂用量大、检测成本高,不利于全面推广应用。超高效液相色谱法(UPLC)是在传统高效液相色谱法(HPLC)基础上开发的一种基于小颗粒填料的液相色谱技术,具分离度好、灵敏度高、分析速度快等优点,在中药特征图谱的研究中发挥了重要的作用[7]。近年来,由于UPLC特征图谱技术突破了单组份定量分析的局限性,能快速、全面、系统地对中药材进行多指标定量、定性分析,因此已逐渐成为中药材质量评价的重要手段[8-12]。本研究建立了基于UPLC同时测定鲜鱼腥草药材特征图谱及5种指标成分含量的方法,显著缩短了分析时间,提高了分析效率,为鲜鱼腥草药材质量评价提供了参考。
1 仪器与材料
1.1 仪器
Waters ACQUITY 型超高效液相色谱系统(包括真空脱气机、二元梯度泵、自动进样器、柱温箱、TUV检测器、Empower数据处理系统,美国Waters公司);Waters液质联用仪(液相部分ACQUITY UPLC H-Class Core System、质谱部分Waters Xevo TQD MS,美国Waters公司);MiliQ Direct 8型超纯水机(德国默克密理博公司);ME203E型分析天平(美国METTLER TOLEDO公司)。
1.2 材料
新绿原酸(批号:wkq18030107,纯度98.0%)、隐绿原酸(批号:wkq16081903,纯度98.0%)对照品均购自四川省维克奇生物科技有限公司;绿原酸(批号:110753-201817,纯度96.8%)、金丝桃苷(批号:111521-201708,纯度95.1%)、槲皮苷(批号:111538-201606、纯度90.6%)对照品均购自中国食品药品检定研究院。色谱级甲醇、乙腈(德国默克股份两合公司);磷酸色谱纯(天津市科密欧化学试剂有限公司);甲醇分析纯(西陇科学股份有限公司)。
16批鲜鱼腥草药材经广东一方制药有限公司魏梅主任药师鉴定为三白草科植物蕺菜(HouttuyniacordataThunb.)的新鲜全草。药材来源见表1。
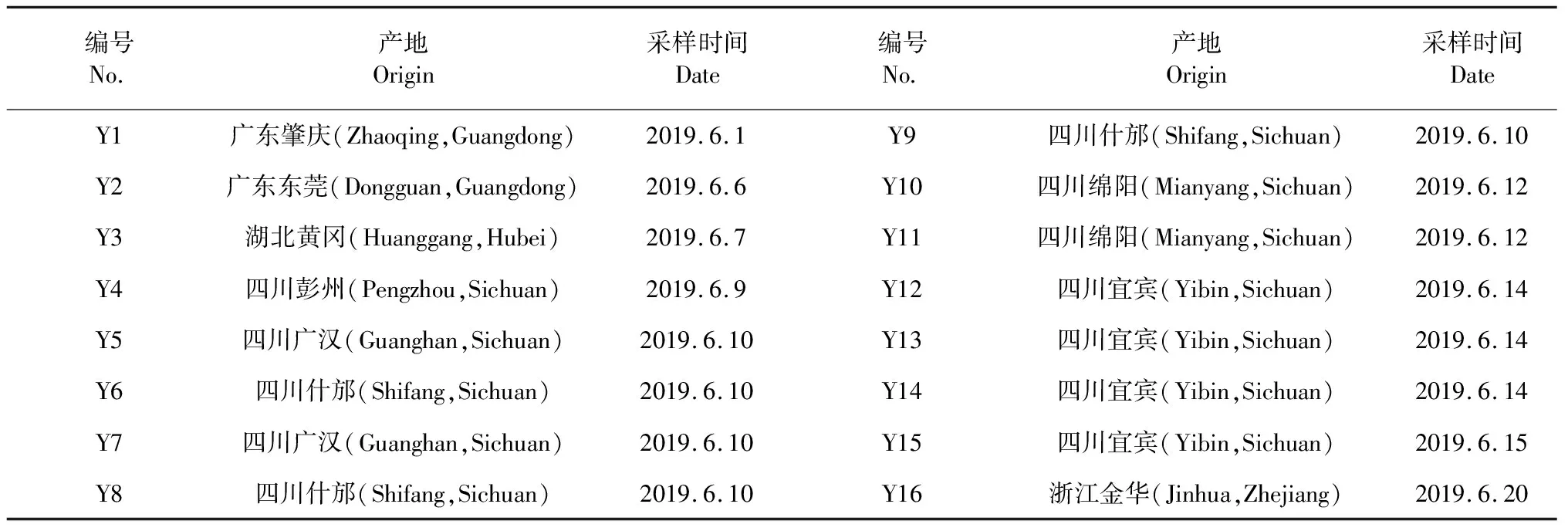
表1 16批鲜鱼腥草药材来源
2 方法
2.1 指标成分含量测定
2.1.1 对照品溶液的制备
取新绿原酸、绿原酸、隐绿原酸、金丝桃苷、槲皮苷对照品适量,精密称定,加90%甲醇溶解、定容,制成质量浓度分别为0.097 12、0.102 87、0.052 04、0.095 14、0.141 88 mg/mL混合对照品溶液,备用。
2.1.2 供试品溶液的制备
取鲜鱼腥草药材约2.0 g,精密称定,置具塞锥形瓶中,精密加入90%甲醇50 mL,密塞,称定重量,加热回流30分钟,放冷,再称定重量,用90%甲醇补足减失的重量,摇匀,过0.22 μm微孔滤膜,取续滤液,即得。
2.1.3 色谱条件
Waters ACQUITY HSS T3(2.1 mm×100 mm,1.8 μm)色谱柱;流动相为乙腈(A):0.1%磷酸水溶液(B),梯度洗脱(0~7 min,5%→11% A;7~10 min,11%→11.5% A;10~13 min,11.5%→20% A;13~20 min,20%→25% A);0~13 min检测波长为326 nm,13~20 min检测波长为254 nm;流速0.3 mL/min;柱温30 ℃;进样量1 μL。
2.1.4 方法学考察
2.1.4.1 线性关系考察
精密吸取混合对照品溶液适量,分别加90%甲醇稀释2、5、10、20、50倍,摇匀。分别按“2.1.3”项下色谱条件进行分析。以峰面积为纵坐标(Y),质量浓度为横坐标(X),绘制各标准曲线,得到线性回归方程和线性范围,结果见表2,新绿原酸、绿原酸、隐绿原酸、金丝桃苷、槲皮苷在各浓度范围内的线性关系良好。

表2 5种指标成分的标准曲线及线性范围

图1 混合对照品(A)、鲜鱼腥草药材样品(B)、阴性对照(C)的UPLC色谱图
2.1.4.2 精密度试验
取Y1号样品按照“2.1.2”项下方法制备供试品溶液,按“2.1.3”项下色谱条件,连续进样6次,计算5种化合物峰面积RSD 值,分别为0.45%、0.56%、1.38%、0.54%、0.54%,表明仪器精密度良好。
2.1.4.3 稳定性试验
取Y1号样品按照“2.1.2”项下方法制备供试品溶液,按“2.1.3”项下色谱条件,分别在制备后0、2、4、8、16、24 h进行检测,计算5 种化合物峰面积RSD 值,分别为1.27%、1.67%、2.62%、1.46%、1.47%,表明样品在24 h内稳定性良好。
2.1.4.4 重复性试验
分别取Y1号样品6份,按照“2.1.2”项下方法制备供试品溶液6份,按“2.1.3”项下色谱条件,分别进样检测,计算5种化合物含量的RSD 值,分别为1.99%、2.99%、2.36%、2.19%、2.94%,表明重复性良好。
2.1.4.5 加样回收率试验
取Y1号样品6份,每份为1.0 g,精密称定,置于具塞锥形瓶中,分别精密加入一定量的对照品溶液,按“2.1.2”项下方法制备供试品溶液,按“2.1.3”项下色谱条件进样检测,计算5种成分的加样回收率,平均回收率依次为97.22%、99.15%、97.43%、100.96%、97.03%,RSD分别为3.78%、3.51%、4.38%、3.24%、3.15%,表明回收率良好。
2.2 特征图谱建立
2.2.1 对照品溶液的制备
同“2.1.1”项下。
2.2.2 供试品溶液的制备
同“2.1.2”项下。
2.2.3 色谱条件
同“2.1.3”项下。
2.2.4 质谱条件
氮气作为质谱离子源的雾化、锥孔气;电喷雾电离正离子模式;毛细管电压:3.0 kV;锥孔电压:25 V;离子源温度:100 ℃;脱溶剂气温度:300 ℃;脱溶剂气流速:800 L/h;扫描时间:0.5 s;扫描时间间隔:0.02 s;扫描范围m/z100~600。
2.2.5 方法学考察
2.2.5.1 精密度试验
取Y1号样品按照“2.2.2”项下方法制备供试品溶液,按“2.2.3”项下色谱条件,连续进样6次,以绿原酸(2号峰)作为参照峰,计算各共有峰的相对保留时间和相对峰面积。各共有峰相对保留时间RSD在0.00%~0.65%范围内,相对峰面积RSD在0.11%~1.00%范围内,表明仪器精密度良好。
2.2.5.2 稳定性试验
取Y1号样品按照“2.2.2”项下方法制备供试品溶液,按“2.2.3”项下色谱条件,分别在制备后0、2、4、8、16、24 h进行检测,以绿原酸(2号峰)作为参照峰,计算各共有峰的相对保留时间和相对峰面积。各共有峰相对保留时间RSD在0.05%~0.60%范围内,相对峰面积RSD在0.31%~1.14%范围内,表明稳定性良好。
2.2.5.3 重复性试验
分别取Y1号样品6份,按照“2.2.2”项下方法制备供试品溶液6份,按“2.2.3”项下色谱条件,分别进样检测,以绿原酸(2号峰)作为参照峰,计算各共有峰的相对保留时间和相对峰面积。各共有峰相对保留时间RSD在0.05%~0.12%范围内,相对峰面积RSD在0.94%~2.58%范围内,表明重复性良好。
3 结果与分析
3.1 样品含量测定
取16批鲜鱼腥草药材,按“2.1.2”项下方法制备供试品溶液,按“2.1.3”项下色谱条件进样检测,测定各批鲜鱼腥草药材新绿原酸、绿原酸、隐绿原酸、金丝桃苷、槲皮苷的含量。结果表明不同批次的鲜鱼腥草药材中5种化合物的含量均有不同程度的差异。

表3 16批鲜鱼腥草药材中5种指标成分的含量
续表3(Continued Tab.3)

编号No.含量Content(mg/g)新绿原酸Neochlorogenicacid绿原酸Chlorogenicacid隐绿原酸Cryptochlorogenicacid金丝桃苷Hyperoside槲皮苷QuercetinY80.3590.1090.0240.1570.607Y90.5350.1630.0250.1720.869Y100.4070.1110.0410.1700.745Y110.5990.1910.0380.4201.210Y120.2460.1270.0640.2550.311Y130.3880.1280.0480.3200.644Y140.3510.1180.0510.2290.540Y150.2670.0650.0250.2920.556Y160.4310.5340.3380.5220.615
3.2 特征图谱的建立及共有峰的鉴定
取16批鲜鱼腥草药材,按照“2.2.2”项下方法制备供试品溶液,按“2.2.3”项下色谱条件进样检测,采用“中药色谱指纹图谱相似度评价系统2012 版”软件对16批鲜鱼腥草药材UPLC特征图谱进行数据处理,建立鲜鱼腥草药材的对照特征图谱(如图2所示),确定共有峰7个,其中采用超高效液相色谱-质谱联用技术(UPLC-MS)以及与对照品对照指认了1、2、3、5、7号峰分别为新绿原酸、绿原酸、隐绿原酸、金丝桃苷、槲皮苷。以绿原酸(2号峰)作为峰1、峰3的参照峰S1,金丝桃苷(5号峰)作为峰4、峰6、峰7的参照峰S2,分别计算各共有峰的保留时间和相对峰面积,结果见表5和6。

表4 基于UPLC-MS的鲜鱼腥草特征图谱共有峰的归属

图2 16批鲜鱼腥草叠加UPLC特征图谱

图3 鲜鱼腥草药材UPLC特征图谱
共有峰相对保留时间和相对峰面积的结果显示,各色谱峰相对保留时间的RSD值范围为0.00%~0.76%,说明7个共有峰在不同鲜鱼腥草样品间的重现性较好,建立的UPLC特征图谱方法较为稳定可行;各色谱峰的相对峰面积的RSD值范围为17.82%~66.73%,波动范围较大,说明不同产地鲜鱼腥草中各成分含量存在差异。
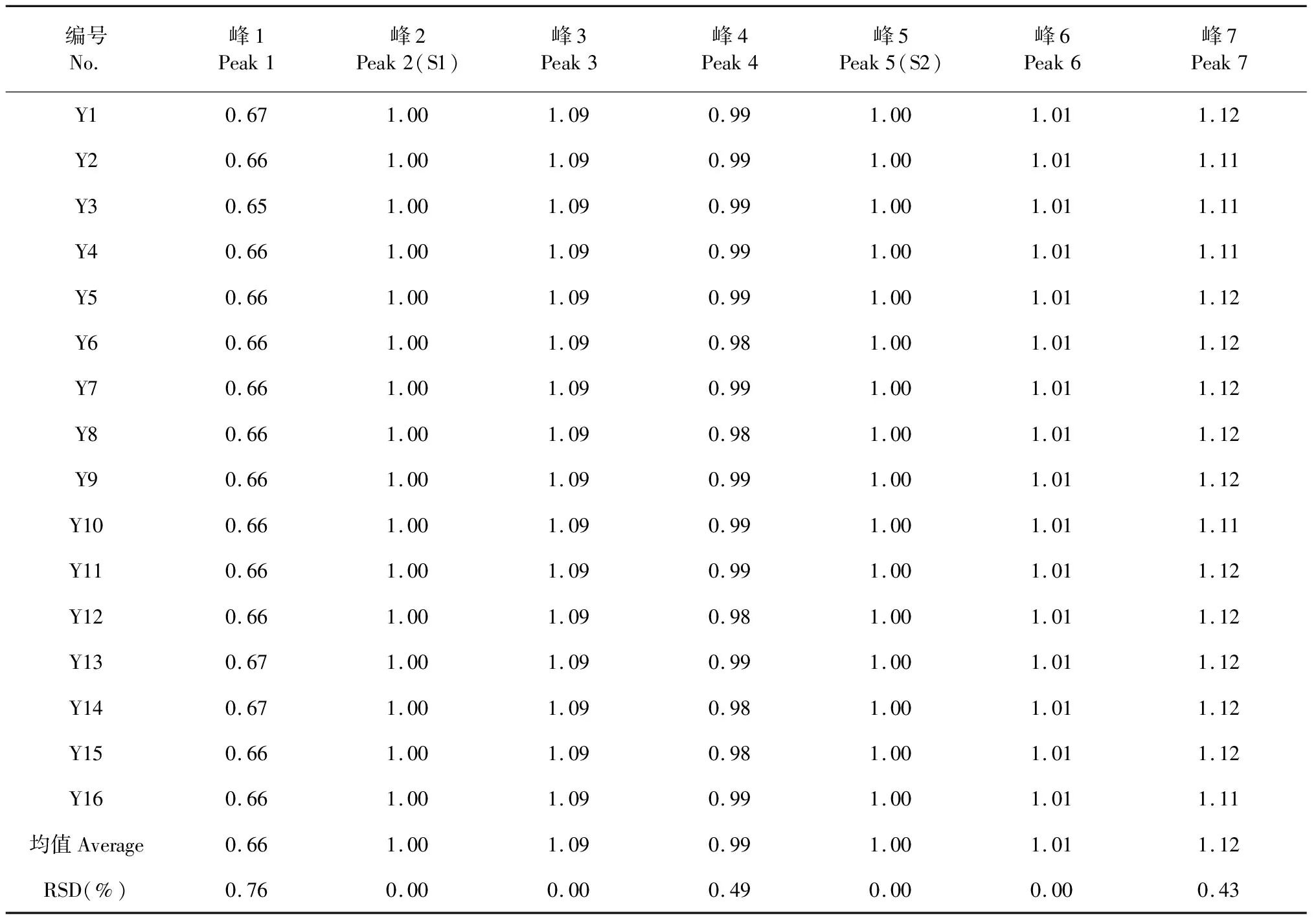
表5 鲜鱼腥草药材共有峰相对保留时间

表6 鲜鱼腥草药材共有峰相对峰面积
3.3 鲜鱼腥草特征图谱的效果评价
3.3.1 相似度评价
采用“中药色谱指纹图谱相似度评价系统2012 版”软件计算16批鲜鱼腥草药材之间以及相对于对照特征图谱(R)的相似度。结果显示,16批鲜鱼腥草样品间的相似度介于0.798~0.999之间;除Y16样品外,其余批次鲜鱼腥草药材样品相对于对照特征图谱的相似度均>0.900,说明鲜鱼腥草药材样品与对照特征图谱间的相似度较好。

表7 鲜鱼腥草特征图谱相似度评价
续表7(Continued Tab.7)

编号No.Y1Y2Y3Y4Y5Y6Y7Y8Y9Y10Y11Y12Y13Y14Y15Y16RY110.9950.8470.9140.9770.9940.9690.9920.9950.9880.9951.0000.9420.9930.9910.9900.8490.991Y120.9350.8970.9890.9430.9350.9380.9500.9330.9200.9190.9421.0000.9740.9730.9560.9480.966Y130.9850.8810.9530.9790.9860.9720.9910.9870.9780.9820.9930.9741.0000.9980.9920.8850.995Y140.9800.8990.9550.9880.9900.9830.9950.9910.9840.9850.9910.9730.9981.0000.9840.8940.999Y150.9900.8220.9270.9510.9720.9420.9740.9740.9600.9730.9900.9560.9920.9841.0000.8530.977Y160.8540.8270.9420.8660.8510.8550.8610.8440.8320.8280.8490.9480.8850.8940.8531.0000.890R0.9780.9060.9490.9940.9940.9890.9980.9940.9900.9880.9910.9660.9950.9990.9770.8901.000
3.3.2 聚类分析
将16批鲜鱼腥草药材特征图谱中的7个共有峰的峰面积相对于称样量进行量化(峰面积/称样量),形成7×16阶数据矩阵,导入SPSS 20.0软件,以离均差平方和法(Ward’s method)为聚类方法、欧氏距离平方法(squared Euclidean distance)为测量距离方法,进行聚类分析。聚类结果如图4所示。
16批鲜鱼腥草大致可聚为3类。广东(Y1和Y2)、湖北(Y3)、四川彭州(Y4)、什邡(Y6、Y8、Y9)、宜宾(Y12、Y13、Y14、Y15)等地的鲜鱼腥草基本可聚为一类;四川广汉(Y5和Y7)、绵阳(Y10、Y11)的鲜鱼腥草可聚为一类;浙江金华(Y16)的鲜鱼腥草单独聚为一类。结果表明,同一产地不同居群的鲜鱼腥草药材质量较为一致,不同产地鲜鱼腥草药材质量存在一定差异。

图4 鲜鱼腥草药材特征图谱聚类分析
3.3.3 主成分分析
利用SPSS 20.0软件对量化后的共有峰峰面积(峰面积/称样量)进行数据标准化处理,标准化后数据见表8。对16批鲜鱼腥草药材的7个共有峰进行主成分分析,计算相关矩阵的方差和特征值,结果见表9、表10。前3个因子累计方差贡献率达到95.797%,特征根大于1,可以代表鲜鱼腥草药材特征图谱中7个共有峰的大部分信息。
对主成分载荷值进行计算,得出各主成分的线型模型,各主成分解析表达式分别为F1=0.131ZX1+0.189ZX2+0.112ZX3+0.214ZX4+0.233ZX5+0.256ZX6+0.209ZX7;F2=-0.210ZX1+0.302ZX2+0.498ZX3-0.160ZX4+0.185ZX5-0.111ZX6-0.314ZX7;F3=0.605ZX1+0.368ZX2+0.081ZX3-0.391ZX4-0.311ZX5-0.119ZX6+0.137ZX7;主成分综合得分函数F=(51.909%×F1+25.809%×F2+18.079%×F3)/95.797%,各批次综合得分结果见表11。

表8 数据标准化

表9 主成分特征值及方差
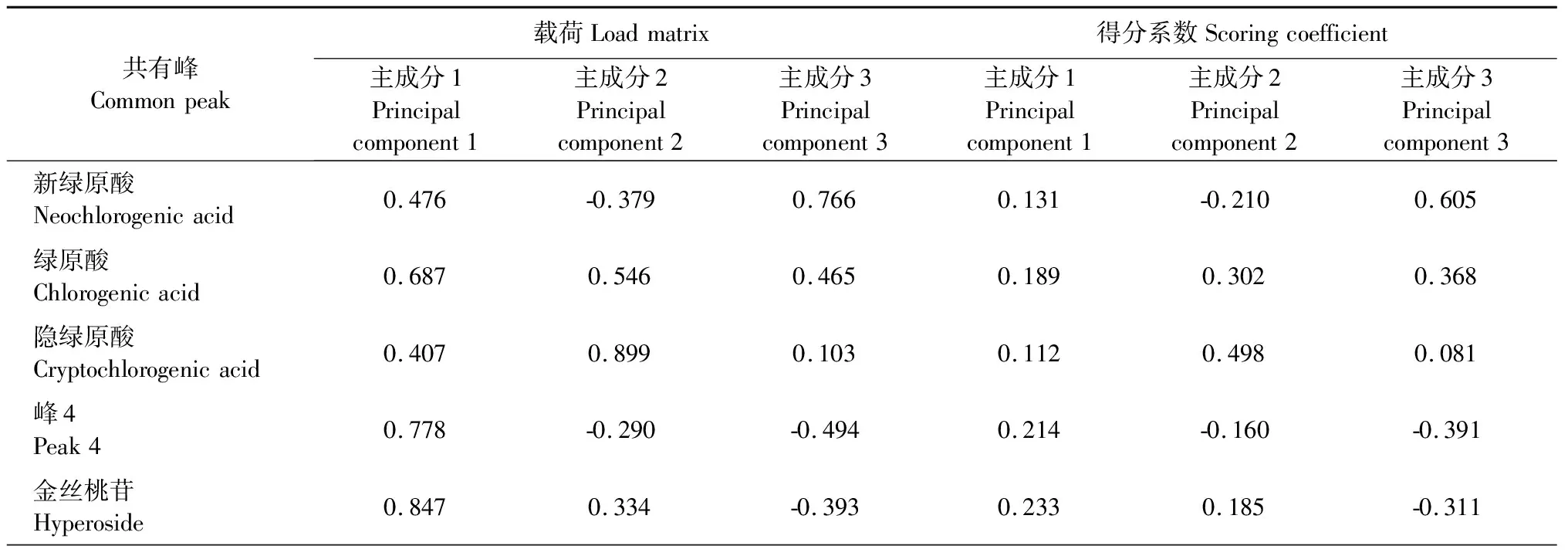
表10 主成分载荷及得分系数矩阵
续表10(Continued Tab.10)

共有峰Commonpeak载荷Loadmatrix得分系数Scoringcoefficient主成分1Principalcomponent1主成分2Principalcomponent2主成分3Principalcomponent3主成分1Principalcomponent1主成分2Principalcomponent2主成分3Principalcomponent3峰6Peak60.932-0.200-0.1510.256-0.111-0.119槲皮苷Quercetin0.761-0.5670.1730.209-0.3140.137
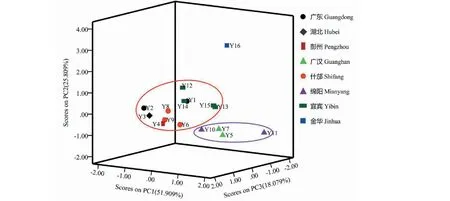
图5 16批鲜鱼腥草主成分得分散点图
主成分得分结果显示,根据主成分分析基本可将不同产地种植的鲜鱼腥草分为3大类。浙江金华的鲜鱼腥草(Y16)单独分为一类;广东(Y1和Y2)、湖北(Y3)、四川彭州(Y4)、什邡(Y6、Y8、Y9)、宜宾(Y12、Y15、Y14、Y15)的鲜鱼腥分为一类;四川广汉(Y5和Y7)、绵阳(Y10和Y11)的鲜鱼腥草分为一类。由此可见,相同产地不同居群的鲜鱼腥草均聚在同一类,16批鲜鱼腥草主成分分析与聚类分析描述的结果基本一致。
利用主成分综合得分函数计算16批鲜鱼腥草的综合得分,结果显示浙江金华种植鲜鱼腥草综合得分远高于其他产地,其次是四川彭州、广汉、绵阳等地,广东、湖北、四川宜宾的鲜鱼腥草综合得分较低,说明不同产地鲜鱼腥草的质量存在较大的差异。
4 讨论
本研究建立了鲜鱼腥草药材质量综合评价的方法,在同一色谱条件下,在20 min内同时完成了鲜鱼腥草5种指标成分含量及特征图谱的测定,极大的缩短了检测时间,降低了分析成本,对于样品的高通量分析具有显著优势,便于推广使用。
本研究分别考察了提取溶剂(50%甲醇、90%甲醇、甲醇、70%乙醇、乙醇)、提取方式(加热回流提取、超声提取)及提取时间(15、30、45、60 min),对鲜鱼腥草药材中5种指标成分提取效率以及特征图谱的影响,结果表明,加入90%甲醇回流提取30 min,提取效率高、效果好。
本研究分别考察了乙腈-0.1%磷酸水溶液、乙腈-0.1%甲酸水溶液、乙腈-0.2%醋酸水溶液3个不同的流动相系统,结果发现采用乙腈-0.1%磷酸水溶液为流动相,各色谱峰分离效果最好。利用PDA检测器对供试品溶液进行全波长扫描,并结合已有的文献研究[6,13,14],分别在254、326、280 nm以及程序波长(0~13 min为326 nm,13~25 min为254 nm)下采集供试品溶液的色谱图,对采集的色谱图进行比较。结果表明,采用程序波长检测方式,色谱峰峰型好,且主要色谱峰吸收强度高,因此选择程序波长检测方式。

表11 16批鲜鱼腥草药材综合得分情况
目前,特征图谱相似度评价法、欧氏距离聚类分析法和主成分分析法等化学模式识别方法在中药材质量、产地差异以及品种鉴别等研究领域的应用已较为广泛[15-17]。本研究引入特征图谱相似度分析、聚类分析以及主成分分析,可以直观衡量不同产地鲜鱼腥草样品间特征图谱变化模式的相似性及亲疏程度。鱼腥草广泛分布在我国南方诸省,市场上以四川、重庆、湖北等地的鱼腥草较为常见,多成分含量测定及特征图谱结果均显示不同产地的鲜鱼腥草药材存在一定差异,引入主成分综合得分函数,可直观且客观地判断各产地鲜鱼腥草药材的整体质量。本研究通过对特征图谱数据进行聚类分析和主成分分析,可将16批鲜鱼腥草分为3类,主成分综合得分结果表明浙江金华种植的鲜鱼腥草质量最优。因此,利用本研究开发的鲜鱼腥草UPLC特征图谱及多指标成分测定的方法,结合综合质量评价函数,可以更全面、直观地评估各产地鲜鱼腥草的质量,规范鲜鱼腥草药材的质量控制。
本研究旨在建立鲜鱼腥草UPLC特征图谱及5种指标成分含量测定的方法,同时结合相关化学识别模式构建鲜鱼腥草药材质量综合评价函数,为鲜鱼腥草质量评价和控制提供参考。由于鱼腥草新鲜样品采集和存储存在一定的困难,本研究收集的鲜鱼腥草样品未能覆盖全国各地,因此关于各产区鲜鱼腥草药材质量评价的结果是否具有更广泛的代表性,还有待于后续进一步扩大样本量系统深入的研究。
