冠状病毒复制分子生物学与防控措施研发进展
2020-06-15张蕾谢建平
张蕾,谢建平
(西南大学生命科学学院现代生物医药研究所,重庆 400715)
新发和再现病原导致疾病爆发是全球性的公共卫生挑战。全球化、人口增加、生境丢失,接触新宿主等因素叠加,动物病毒跨种传播对公共卫生的威胁尤其突出。其中,冠状病毒(Coronavirus,CoV)因为导致严重新型冠状病毒肺炎(Novel coronavirus pneumonia,NCP)的新型冠状病毒2019-nCoV、导致严重急性呼吸综合征相关冠状病毒SARS-CoV(severe acute respiratory syndrome CoV)和中东呼吸综合征相关冠状病毒MERS-CoV(Middle East respiratory syndrome CoV)等备受关注。
冠状病毒1937年最初从鸡的感染组织中发现。冠状病毒高度流行、分布广泛,其基因组遗传多样性巨大(表1)、重组频繁,加之人-动物界面接触增加,尤其是食用野生动物,频繁的跨物种感染和偶尔的溢出事件,新冠状病毒有可能在人群中周期性出现。已知有七种冠状病毒会引起人类疾病。除上述三种,另外四种冠状病毒-人冠状病毒(Human coronaviruses,HCoVs)-229E、HCoVOC43、HCoV-NL63和HCoV-HKU1,在免疫功能正常的个体中普遍存在,一般呈现普通感冒症状。其中HCoV-NL63、HCoV-229E和HCoV-OC43来自蝙蝠。冠状病毒家族中鼻病毒是普通感冒的主要原因(约10%~30%)。有些冠状病毒是家禽和家畜的重要致病菌,可引起呼吸道、肠道、肝脏和神经系统疾病。如鸟支气管炎病毒(Avian Infectious Bronchitis Virus,AIBV),猪肠胃炎病毒(Transmissible Gastro-Enteritis Virus,TGEV),猪流行性腹泻病毒(Porcine epidemic diarrhea virus)变异株,γ属的禽传染性支气管炎病毒(Infectious bronchitis virus,IBV),猪急性腹泻综合征(SADS)等,这些病毒多次给世界畜牧业造成巨大损失。

表1 部分冠状病毒及其宿主细胞受体
冠状病毒也成为研究病毒-宿主相互作用的模式病毒。一般发生不明原因肺炎时,如果怀疑是冠状病毒,首选需要通过测序、细胞培养分离等方法确定病原,为后续病毒溯源、病毒致病机理、动物模型建立、疾病发生、发展和传播规律及临床诊治、传播力大小、快速检测技术和产品研发、抗病毒应急药物和抗体类药物、疫苗研发等相关研究奠定基础。目前的测序技术和大数据可以快速锁定病原。比如新冠肺炎致病菌的锁定只用了一周左右。但是,因为缺乏动物模型,目前很难确定冠状病毒感染致病的关键因子。
关于冠状病毒的研究较多。截至2020年2月7日下午7点,本文研究利用coronavirus在NCBI genome中检索到40个冠状病毒基因组。其中最早的为1993年。截止2020年2月7日,用coronavirus检索NCBI PubMed发现收录文献14673篇。用“冠状病毒”检索中国CNKI期刊库发现4546篇,其中综述195篇。2003年是世界上冠状病毒研究的分水岭。2003年以前,每年最多发表文章89篇,2003年以后,每年一般发表130篇以上。主要原因是2003年的非典型肺炎(SARS)和2012年的中东呼吸综合征(Middle East respiratory syndrome coronavirus,MERS-CoV),以及导致最近“新冠状病毒肺炎”的2019新冠状病毒(2019 novel coronavirus,2019-nCoV)疾病高度流行。研究中比较重要的进展有:以SARS-CoV作为模式,综合系统生物学和反向遗传学技术,构建动态转录网络(Dynamic transcriptional network),寻找疾病相关表型的宿主或病毒遗传因子,以及探索人群水平的致病机理。
1 冠状病毒基因组特征和主要宿主
冠状病毒(图1)属于巢病毒目(Nidovirales)、冠状病毒科(Coronaviridae)、正冠状病毒亚科(Orthocoronavirinae)、冠状病毒属(Coronavirus),是自然界广泛存在、物种多样的一大类有包膜、不分节段的单股正链RNA病毒,基因组全长27~32kb,螺旋状核衣壳结构外面包裹有脂双层,可以感染人、牛、鼠、猪、猫、犬、雪貂、骆驼、兔子、蝙蝠与禽类等宿主。目前有α、β、γ和δ四个属。β冠状病毒是包膜性病毒。新型肺炎病毒2019-nCoV就属于冠状病毒科β冠状病毒属,与一种来自蝙蝠的冠状病毒的序列一致性高达96%。该病毒经飞沫、气溶胶、接触等途径传播,潜伏期感染者也具传播性。目前数据表明:2019-nCoV比SARS-CoV毒力弱,但传播力强。可能2019-nCoV与SARS冠状病毒使用相同的受体-血管紧张素转化酶2(ACE2)进入宿主细胞。病毒S蛋白结合宿主细胞受体的能力是CoV在新物种中存活的关键,但不是唯一因素。
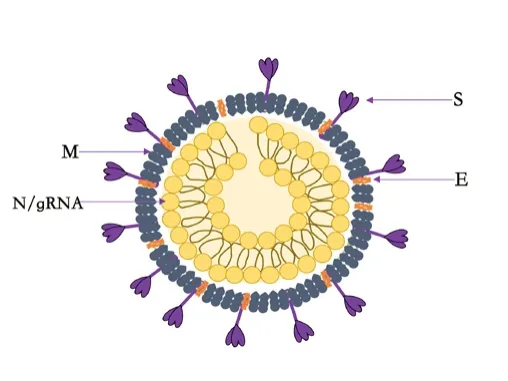
图1 冠状病毒形态示意图(2019-nCoV)
一般RNA病毒的基因组都尽可能小,以避免错误灾难(Error catastrophe)。CoV作为巢病毒目中基因组最大的成员,克服错误灾难的方法是产生一个大的复制复合体(Replication complex)。该复合体包括非结构蛋白(Nonstructural protein,nsp)nsp14的3'-5'核酸外切酶(Exoribonuclease)活性介导的校读功能,具有RNA合成和修饰活性。这个巨大、复杂的RNA复制机器允许CoV基因组达到32kb,还能够存活。冠状病毒依赖RNA的RNA聚合酶(RNA dependent RNA polymerase,RdRp)的忠实性降低,突变更快,尤其是群体大,代时短,因此突变率高,遗传变异大,与宿主细胞表面受体如二肽基肽酶4(Dipeptidyl peptidase 4,DPP4)相互作用的病毒S蛋白的突变、重组、高突变子位点(Mutator alleles),突变稳健性(Mutational robustness)等因素叠加使得冠状病毒的宿主范围不断扩大。根据选择压力,CoV蛋白可以分为三组:刺突蛋白、保守性蛋白和可变蛋白。新CoV出现,这三组蛋白的功能必需协调,既维持病毒关键功能,也克服种障碍。
以2019-nCoV为例(图2),透射电子显微镜下,负染色的2019-nCoV粒子通常是球形,具有多形性。直径60~140nm。人气道上皮超薄切片可见胞外游离病毒颗粒和胞质膜结合小泡内充满病毒颗粒的包涵体[1]。
冠状病毒的宿主很广泛[2]。目前发现与人类肺炎关系密切的宿主都涉及蝙蝠。蝙蝠是包括冠状病毒在内的多种病毒重要宿主。人类聚居区附近携带病毒的蝙蝠容易将病毒传播给人类和牲畜。这主要是蝙蝠进化历史悠久、地理多样性强,季节性迁徙,物种多样性和丰度高(大约占哺乳动物多样性的20%)、种群密度大、具有独特免疫、生理等特征。蝙蝠携带多种CoV。目前,关于蝙蝠不发病的假说主要有以下几种:(1)蝙蝠飞行产生大量活性氧(Reactive oxygen species,ROS),活性氧调控基因表达,限制氧化胁迫,抑制病毒复制和致病。(2)蝙蝠多样性的病毒库可能与其独特的先天免疫有关。蝙蝠因为缺乏炎症体途径的Pythin(PYRIN and HIN domain-containing)和天然杀伤免疫球蛋白样受体KIR(natural killer immunoglobulin-like receptors)基因,或者极低的表达,病毒感染导致的疾病和损伤有限。蝙蝠组成性表达某些亚型的干扰素,既可以限制发病,也允许病毒低剂量存在。(3)病毒和蝙蝠可能是共生关系。病毒可能是蝙蝠微生物组中激发免疫的关键,类似人的疱疹病毒。另外,就疾病和适应性免疫而言,肠道感染和呼吸道感染还是不同。病毒嗜性因物种和组织而异,这可能也是蝙蝠不发病的原因[3]。蝙蝠具有完整的适应性免疫系统,肠道的病毒导致适应性免疫强度较低,病毒可以维持在类似人微生物组成员滴度的水平。(4)蝙蝠有些因素可以维持病毒的存在,也促进了CoV准种(quasispecies)库的多样性。蝙蝠飞行过程中,可能短期积累ROS。ROS的突变效应可以超过病毒聚合酶的校读修复能力,改变病毒聚合酶忠实性,提高病毒物种多样性,为跨种传播提供基础。蝙蝠连续产生I型干扰素可能选择有利于突变的病毒物种,增强病毒对新宿主天然免疫的抗性,跨种传播后复制更有优势。蝙蝠缺乏关键的炎症介导因子,没有降低这些免疫应答的选择压力。病毒感染新宿主后,新宿主产生大量病理性炎症应答,这与在MERS-CoV和SARS-CoV感染人后观察到的现象吻合。蝙蝠中大量病毒准种存在的因素,也使得病毒具有多样性,在新物种中出现。
2 冠状病毒复制、包装和与宿主相互作用的过程与关键分子[4-7]
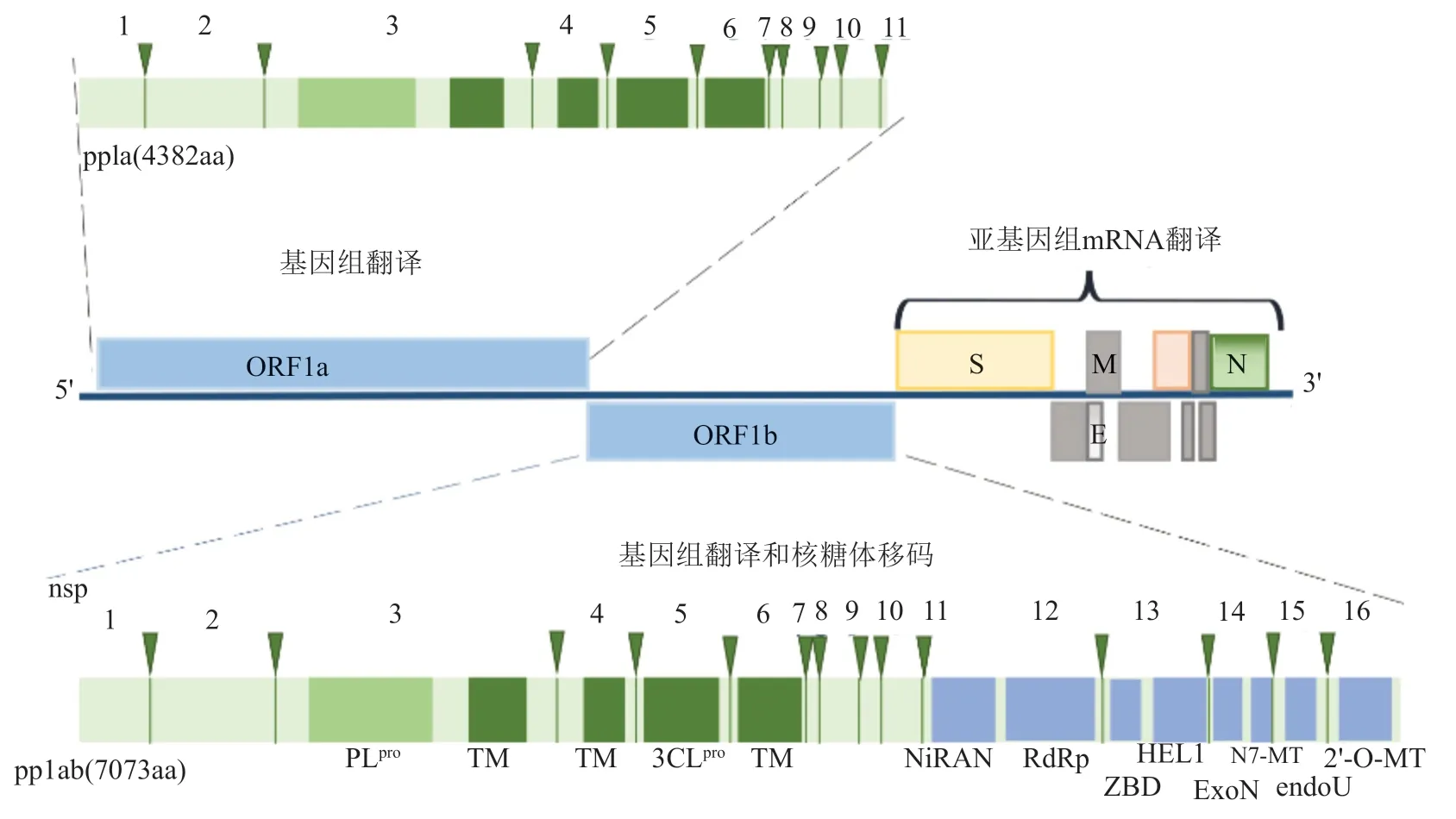
图2 冠状病毒基因组结构示意图(以SARS-CoV为例)
认识病毒复制、病毒-宿主相互作用是解决冠状病毒防控措施的关键。冠状病毒比其他正链RNA病毒的基因组大,基因表达策略复杂,蛋白质组也大,与宿主相互作用复杂。病毒在宿主细胞中表达的蛋白质,一方面用于病毒复制或者组装,另一方面与宿主相互作用。为病毒复制提供更佳环境、改变宿主基因表达、拮抗宿主抗病毒策略。冠状病毒蛋白质组复杂,复制过程复杂。对于宿主因子在冠状病毒复制中的作用、细胞中病毒RNA合成机器的组装与功能,逃避宿主细胞先天免疫应答的相关研究较少。
2.1 核酸合成酶
冠状病毒基因组编码两个具有RNA依赖的RNA合成酶(RNA-dependent RNA polymerase,RdRp)活性的蛋白,即nsp12与nsp8,前者主要是利用引物合成长链的基因组或者mRNA,后者利用RdRp活性合成短链RNA,作为引物供前者合成长的RNA链,nsp8是冠状病毒基因组合成起始阶段的关键。
nsp14,ExoN与nsp10(在冠状病毒中相对保守)形成复合体,介导3′-5′核酸外切酶(Exoribonuclease)活性,类似DNA聚合酶的校读活性,具有DEDD 超家族。nsp14 DEDD突变后,突变率提高15~20倍。nsp10-nsp14复合体提高复制忠实性。基因重组、基因重复、旁系基因进化、利用重叠读框从头产生等都可以获得基因。尚待明确基因组扩增、附属基因是否有利于冠状病毒扩大宿主。
RNA病毒进行基因组选择性包装。这是病毒侵入和破坏宿主先天免疫系统的关键。许多包装方式都涉及基因组包装信号。对冠状病毒基因组包装的认识主要来自两个病毒:小鼠肝炎病毒(Mouse hepatitis virus)和传染性肠胃炎病毒(Transmissible gastroenteritis virus)。冠状病毒第一个包装信号发现了30多年,但对其功能认识仍然不足。目前关于包装信号研究的结论尚无一致意见。争议主要体现在包装信号识别过程中病毒核衣壳蛋白或膜蛋白是否发挥主要作用。
冠状病毒具有独特的非连续转录模式。基因组RNA 5′末端包括一个末端帽子结构,其后是65~98个核苷酸大小的前导序列以及200~400个核苷酸大小的不译区,3′末端则包括200~500个核苷酸大小的不译区和poly(A)尾。冠状病毒基因组RNA具有mRNA功能,有感染性。冠状病毒是正链RNA病毒(Positivestranded RNA(正链RNA)viruses),特殊之处是基因组大,约30kb。其基因组结构为多顺反子,利用独特机制产生嵌套的亚基因组(Subgenomic,sg)mRNAs,用于表达位于复制酶(Replicase)ORFs 1a和1b下游的编码结构蛋白和辅助蛋白可读框(Open reading frames,ORFs)(图3)。sg mRNAs结构特点是共3′末端,也含有共同的5′前导序列。sg RNAs的前导序列和结构序列在不连续的负链RNA合成中连接,提供每个sg mRNAs的亚基因组长度的模板。
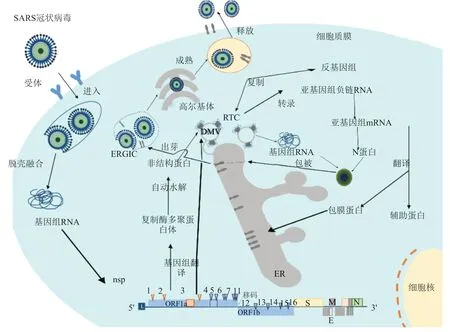
图3 冠状病毒复制和感染示意图(以SARS-CoV为例)。
冠状病毒复制酶基因转录的非结构蛋白具有多种功能,如多聚蛋白加工、复制酶复合体的形成、病毒缺陷复制、RNA转录、翻译和蛋白合成、加工和修饰、感染宿主、结合宿主蛋白质、调控毒力和疾病轻重等。深入研究非结构蛋白有助于了解病毒的致病机制,为开发新型抗病毒药物和研究病毒检测和防控方法提供靶点。SARS-CoV、IBV、HCoV229E等冠状病毒的非结构蛋白高度保守,研究SARS-Cov或IBV非结构蛋白将有助于了解冠状病毒。
复制与细胞膜组分密切相关,尤其是内质网(Endoplasmic reticulum,ER)。被其感染的细胞呈现内质网胁迫,诱导未折叠蛋白质应答(Unfolded protein response,UPR),关闭全局性翻译,增加内质网折叠能力,试图恢复内质网稳态。内质网胁迫过长,UPR会诱导细胞凋亡。冠状病毒-宿主相互作用涉及内质网胁迫和UPR。UPR涉及三条途径,任何一条途径被激活,如丝裂原活化蛋白激酶(Mitogenactivated protein(MAP) kinase)、自噬或者先天免疫应答。病毒复制和致病可能得益于ER胁迫和UPR。不同凋亡途径之间相互作用。
病毒粒子组装和出芽:包膜病毒中,冠状病毒特殊之处在于病毒包膜组装发生在ERGIC。病毒粒子自此出芽进入腔,再进入宿主分泌途径,最后出胞。M蛋白协调病毒组装,病毒样颗粒(svirus-like particle,VLP)的产生和释放需要M和E。缺乏E蛋白编码基因(ΔE)的重组冠状病毒(Recombinant CoVs,rCoVs)形态异常。MHV E蛋白C-端残基变为丙氨酸后,病毒粒子变得对温度敏感,更长,而不是典型的球状。病毒斑形态异常,小且不规则,边缘锯齿状。重组SARS-CoV-ΔE(Recombinant SARS-CoVΔE,rSARS-CoVΔE)成熟病毒粒子减少,含致密颗粒物质的小泡增多。这可能是病毒组装失败,病毒粒子不成熟。CoV E viroporin具有阳离子选择性,优先通过单价的Na+和K+。合成SARS-CoV E类似viroporin,能够运输Na+、K+和Cl-,对此显示对Na+比K+选择性强,对Cl-选择性很弱。
核酸内切酶(Endoribonuclease,EndoU)[8-10]:由非结构蛋白(Nonstructural protein,nsp)nsp15编码,nsp15具有尿苷酸特异性核酸内切酶(U-specific endoribonuclease,NendoU)活性,327~375个氨基酸,38~42kDa,其N端参与蛋白之间的工作,C端包含酶活性区域,其活性区域在所有冠状病毒中均保守。nsp15发挥活性依赖与Mn2+结合。它可作用于单链和双链RNA,切除3'端GUU或GU序列的尿苷酸, 形成2'-3'三磷酸二酯环末端。Mn2+可激发酶活性,nsp15-Mn2+结合作用较弱,但这种结合可使蛋白构象发生显著变化。最早认为是巢病毒目病毒复制复合体的组分。后来发现,nsp15缺陷的冠状病毒在成纤维细胞中,也可以复制和存活,滴度甚至与野生型无异。EndoU介导病毒双联RNA逃避宿主巨噬细胞中传感器的识别。病毒nsp15/EndoU在致病中的作用,以及作为药物和疫苗设计的靶标。
CoV E蛋白(Envelope,E):76~109个氨基酸,分子量8.4~12kDa,膜整合蛋白,参与冠状病毒生活史如病毒组装、出芽、包膜形成和致病。对其结构模体、拓扑结构、离子通道(viroporin),与病毒及宿主细胞蛋白质的研究较多。冠状病毒E蛋白经历多种蛋白质翻译后修饰。其中IBV、SARS-CoV、MHV和CoV E蛋白被棕榈酰基化。而棕榈酰基化的靶标是TMDs附近的半胱氨酸残基。即MHVA59 E蛋白半胱氨酸双突变或者三突变为丙氨酸后,形成VLP的能力降低。验证MHVE蛋白三突变后不稳定,容易降解,突变菌株的产量降低,说明E蛋白在MHV病毒组装中必不可少。提示IBV E蛋白棕榈酰基化不影响定位到高尔基体,因为半胱氨酸突变的E蛋白与野生型蛋白,在定位方面无差异。证明SARS-CoV E可以被泛素化,但是功能未知。SARS-CoV nsp3与E共定位,nsp3 N-端泛素样功能域-1介导其相互作用。E蛋白可以被泛素化,而且泛素化状态与稳定性和半衰期负相关。SARS-CoV辅助蛋白8b表达较晚,可能下调E蛋白产生,调控病毒的产生,维持病毒滴度。IBV E蛋白腔N-端有一个糖基化位点,SARSCoV E预测有两个糖基化位点。寡糖与保守序列Asn-X-Ser/Thr上的天冬酰胺残基连接(N-连接糖基化)。糖基化修饰有利于招募宿主分子伴侣如钙连蛋白和钙网蛋白,帮助病毒或者宿主细胞蛋白质正确折叠和运输。S蛋白和E蛋白具有三半胱氨酸模体[11]。即E蛋白氨基末端后(NH2- … L-Cys-A-Y-Cys-Cys-N …-COOH)以及S蛋白C-端(NH2- … S-CysG-S-Cys-Cys-K … -COOH)。这些模体可能是E和S相互作用的基础,形成二硫键。CoV结构蛋白中,M和E蛋白相互作用的研究比较透彻。共表达M和E足以形成和释放VLP。介导相互作用的是两者的C-端,相互作用位于ERGIC细胞浆一侧。如果耗尽这些功能域,VLPs产量迅速降低。
CoV E蛋白可以相互作用,寡聚化,形成病毒离子通道-病毒孔蛋白。这与TMD有关。合成对应SARS-CoV E TMD的肽段,可以形成二聚体、三聚体、五聚体。与此相关的残基为天冬酰胺15(N15)、缬氨酸25(V25)。缺乏E的CoV可望作为疫苗。
有些CoVs形成完整、有感染性的病毒粒子,不需要全部结构蛋白。这也提示有些结构蛋白的功能可能冗余,或者具有结构蛋白之外的功能,比如参与病毒复制。S蛋白介导病毒与宿主细胞表面受体黏附、病毒和宿主细胞膜融合,有利于病毒进入宿主细胞。同时,也可能介导与邻近未感染细胞融合。病毒在细胞之间扩散可能与形成巨大的多核合胞体(syncytia)有关,破坏中和病毒的抗体。N蛋白主要结合CoV RNA基因组,形成核衣壳。N定位到内质网-高尔基体(Endoplasmic reticulum (ER)-Golgi region)与其组装和出芽有关。瞬间表达N蛋白可以大量增加有些CoVs产生病毒样颗粒(Virus-like particles,VLPs),提示N蛋白在包膜形成中的功能,但与完整病毒粒子形成的相关性不大。SARS-CoV核衣壳蛋白N干扰宿主RIG-I泛素化。MERS-CoV和SARS-CoV蛋白N特异性表位诱导呼吸道记忆CD4+T细胞,具有免疫保护功能。M蛋白丰度最高,但是负责确定病毒包膜的形状,是CoV组装的核心组织者,与冠状病毒的其他结构蛋白相互作用。S蛋白与M蛋白相互作用,是将S蛋白滞留在内质网-高尔基体过渡空间/高尔基复合体(ER-Golgi intermediate compartment,ERGIC)/Golgi complex,以及掺入病毒粒子所需,但S蛋白不是病毒粒子组装所必需。M与N结合,稳定病毒粒子核衣壳,以及病毒粒子内部核心,最后完成病毒组装。病毒的多数蛋白位于胞内运输途径即内质网、高尔基体和ERGIC,参与CoV组装和出芽。E在病毒繁殖和组装中发挥作用。缺乏E的重组CoVs病毒滴度明显减少,病毒成熟受损,子代不完全。
冠状病毒可以利用宿主的泛素化与去泛素化修饰系统,甚至编码自己的泛素/去泛素酶,调控蛋白质翻译后修饰,驱动病毒生活史。MERS-CoV nsp3去泛素酶抑制宿主免疫应答。nsp3本来防止ADP核糖基化的大功能域突变后,病毒的毒性降低。S-腺苷甲硫氨酸(S-adenosyl-L-methioline,SAM)与nsp16(2-O-甲基转移酶)结合,促进MERS-CoV复制,招募来更多别构激活因子在冠状病毒中相对保守的nsp10。
2.2 与冠状病毒基因组复制或者基因表达相互作用的宿主因子
相对于其他正链RNA病毒,参与冠状病毒复制的宿主因子的研究甚少。与冠状病毒进入细胞有关的宿主受体研究提示冠状病毒复制循环的第一步是进入靶细胞,如气管上皮细胞,特别是纤毛上皮细胞和肺泡II型肺细胞。其他与致病有关的包括浸润免疫细胞如巨噬细胞、嗜中性粒细胞、树突状细胞(Dendritic cell,DC)。其中DC还可以进一步分为髓型DC(Myeloid-type DC,mDC)和浆型DC(pDC)。两种DC都是清除病毒的关键。激活的pDC产生大量I型IFN,提示附近细胞有感染。mDC产生I型IFN量不如pDC,可以分泌激活B细胞和T细胞需要的趋化因子,在适应性免疫中非常关键。感染冠状病毒S糖蛋白与宿主细胞表面的蛋白质受体识别并结合。其中冠状病毒S蛋白是1型糖蛋白,1160~1400个氨基酸,由S1和S2亚基组成,在病毒粒子表面是三聚体,furlin蛋白负责在裂解位点RRXRR处切开S1和S2。S1区参与受体结合,含有可以作为受体结合功能域(Receptor-binding domain,RBD)的N-和C-端功能域(S1-NTD和S1-CTD),提示决定细胞嗜性(Cell tropism)的主要功能在S1-CTD。S2区形成刺突三聚体的茎,激发病毒包膜和靶细胞膜融合。S1-NTD与宿主细胞表面聚糖(Glycan)相互作用,有利于病毒结合和进入。基于betacoronavirus S1-NTD晶体结构,以及其他冠状病毒S1-NTDs的序列保守性,所有冠状病毒S1-NTDs具有半乳糖凝集素折叠,介导结合唾液酸如N-糖基神经氨酸(N-glycolylneuraminic acid,Neu5Gc),N-乙酰神经氨酸(N-acetylneuraminic acid,Neu5Ac)和/或5-N-乙酰-9-O-乙酰神经氨酸 (5-N-acetyl-9-Oacetylneuraminic acid、Neu5、9Ac2) 。例外是小鼠肝炎病毒(Murine hepatitis virus,MHV)S1-NTD,结合癌胚抗原相关细胞黏附分子1(Carcinoembryonic antigen-related cell adhesion molecule 1,CEACAM1)的N-端D1功能域,免疫球蛋白超家族的I型膜蛋白。进入细胞,大多数alphacoronavirus的S1-CTDu氨肽酶N(Aminopeptidase N,APN,也称为CD13)相互作用。但alphacoronavirus HCoV-NL63利用另一个I型糖蛋白血管紧张素转化酶2(Angiotensin-converting enzyme 2,ACE2)。ACE2是金属蛋白酶,属于I型跨膜糖蛋白,具有羧肽酶活性,在调节心、肾功能以及控制血压中起关键作用。人类ACE2的细胞外区域由2个亚基组成,其中锌金属肽酶区域可以进一步分成2个亚域(I和II),形成一个长而深的裂缝,ACE2的N-端细胞外功能域有两个alpha-helical。ACE2也是动物betacoronavirus SARS-CoV的受体。人、哺乳动物与鸟类ACE2分子仅少数位置氨基酸有差异,研究这些细微差别是冠状病毒宿主特异性以及种间跳跃突变的重要线索。SARS-CoV RBD中Asn479、Thr487位的氨基酸对RBD-ACE2的亲和力至关重要,其突变能够调控RBD与不同宿主ACE2的结合力。betacoronaviruses MERS-CoV和蝙蝠HKU4利用另外一个二肽基肽酶4(属于丝氨酸蛋白酶家族,Dipeptidyl peptidase 4,DPP4,也称CD26)作为受体。DPP4广泛分布于上皮组织和内皮组织内部,通常通过裂解激素和趋化因子的N末端肽键而改变其活性,改变机体或细胞的葡萄糖代谢、免疫应答、黏附和凋亡等过程。MERS-CoV S蛋白对人DPP4亲和力高,HKU4 S蛋白结合DPP4亲和力更高。肽酶抑制剂不影响病毒进入,提示SARS-CoV和MERSCoV受体和进入不依赖受体的肽酶活性,只依赖结合特定宿主受体。DC-SIGN(Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin)是巨噬细胞和树突状细胞表达的C型凝集素,也称CD209,可以识别常见的病毒及细菌的高度甘露糖糖基化分子。冠状病毒的S蛋白高度糖基化,可以结合DC-SIGN。猫传染性腹膜炎病毒(Feline infectious peritonitis virus,FIPV)也属于冠状病毒,感染单核细胞和巨噬细胞后导致全身扩散。表达外源性DCSIGN的非易感细胞可能变得易感。I型和II型FIPV都能以DC-SIGN为共受体或替代受体,且不依赖唾液酸。
胞外细胞表面结合和/或溶酶体蛋白酶在冠状病毒进入宿主细胞中发挥作用,因为其激活S蛋白的融合活性。细胞表面结合的跨膜丝氨酸蛋白酶2(Cell surface-associated transmembrane protease,serine 2,TMPRSS2)切割S蛋白,激发SARS-CoV与细胞膜融合。TMPRSS2也是切割和激活HCoV-229E和MERSCoV S蛋白所需条件。内吞过程,早期内吞体(Early endosome)中的SARS-CoV S蛋白被溶酶体组织蛋白酶L和P切割,病毒包膜与内吞体膜融合,病毒RNA释放到被感染细胞的细胞浆。过程与MERS-CoV进入细胞的机理类似。抑制细胞蛋白酶furin后,MERS-CoV不能进入细胞,但是SARS-CoV能够进入,提示furin介导的切割是MERS-CoV有效进入的关键。MHV A59与晚期溶酶体融合,切割S蛋白依赖晚期溶酶体中较低的pH。不同冠状病毒为何分别与早期或者晚期溶酶体融合,以及宿主嗜性和致病性的关系,目前尚不清楚。S蛋白切割机理也比较复杂。MERS-CoV进入依赖产病毒细胞中furin介导的切割,切割后的MERS-CoV S蛋白被受体细胞的蛋白酶加工,病毒粒子经内吞体进入细胞,或者与细胞浆膜融合。含有未切割S蛋白的MERS-CoV病毒粒子则与晚期溶酶体融合。
宿主蛋白酶是多种B型病毒进入宿主细胞的重要屏障。绕过该屏障可使B型病毒通过未知受体进入人类细胞。冠状病毒S蛋白与宿主细胞表面受体的结合是宿主嗜性的关键决定因素。SARS-CoV S蛋白的RBD的少数突变(N479L和T487S)足以影响与人ACE2的亲和力。MERS-CoV S蛋白较蝙蝠HKU4 S有两个突变,则可以结合人DPP4受体,但不能介导进入,因为不能被人蛋白酶激活。MERS-CoV S蛋白的两个突变(S746R和N762A)可以被人蛋白酶切割,病毒可以进入人细胞,这可能与动物转移MERS-CoV有关。
几个株系的乙型冠状病毒表面携带血凝素-酯酶(Hemagglutinin-esterase,HE)。HE蛋白含有凝集素结合功能域,介导与O-乙酰化唾液酸结合,也具有唾液酸-O-乙酰酯酶受体破坏酶活性,防止黏附到非许可细胞,HE也有利于结合主要受体后进入靶标细胞。
2.3 冠状病毒复制需要修饰宿主细胞膜
正链RNA病毒的RNA合成发生在细胞浆,结合病毒诱导的细胞内膜派生结构。已知正链RNA病毒诱导的膜复制相关结构有两种。一种是单层膜结构,在细胞器如内质网、过氧化物酶体、内吞体的膜上形成一个向内弯曲的小球,具体是哪种细胞器,视病毒类型不同而异。病毒复制机器位于这些小球内,RNA产物通过连通小球内侧和细胞浆的通道向外输送,参与翻译或者组装病毒颗粒组装。黄病毒和甲病毒诱导形成这种单层膜的复制细胞器。另一种是双层膜小泡(Double-membrane vesicles,DMVs),一般伴随其他结构如小管、折叠的内质网或者卷曲的膜,一起在细胞浆形成网状管系统。小RNA病毒、动脉炎病毒和黄病毒样丙肝病毒诱导类似结构。病毒nsps具有跨膜区,或者以其他方式锚定到膜上,驱动形成这些结构。冠状病毒的nsp3、nsp4和nsp6参与形成这些结构。
关于参与冠状病毒复制细胞器形成的宿主因子和途径的假说和数据很多,但尚无定论。内质网膜参与是目前普遍接受的。病毒诱导的双层膜结构表面或者内部具有核糖体和内质网标记分子如sec61α和蛋白质二硫键异构酶(Protein disulphide isomerase,PDI)也支持这个观点,冠状病毒诱导的DMV外膜与内质网池(ER cisternae)是一个连续体。鉴于与ER的联系,有人提出包括依赖外被体蛋白(Coatomer protein,COP)及相关因子如GBF-1途径的分泌途径在复制中发挥重要作用。目前尚未找到冠状病毒诱导的膜结构的标记分子nsp4和分泌途径标记分子的共定位证据。siRNA沉默筛选发现COPB2(β′-COP)抑制SARS-CoV复制。同一机器的组分COPB1和GBF1也严重影响SARS-CoV复制,证实分泌途径的完整性对于病毒复制非常关键。磷脂酰肌醇4-激酶(Phosphatidylinositol 4-kinases,PI4K)对于正链RNA病毒复制非常重要。PI4K等激酶被招募到膜修饰位点,刺激产生PI4P脂类,支持形成病毒复制结构和功能发挥。PI4K异构体PI4KIIIbeta对SARS-CoV复制非常重要。其发挥功能主要是在病毒入侵细胞,而不是复制的后期。病毒的脂类囊膜可能与宿主细胞膜通过相溶性进行扩散。自噬体(Autophagosome)为双层结构。冠状病毒诱导形成双层结构,提示自噬可能被冠状病毒劫持来进行复制。但是,有实验表明自噬因子Atg5并非冠状病毒在原代细胞中复制所需。ERED,膜运输和自噬等可能都参与冠状病毒复制。
2.4 冠状病毒调控被感染细胞的翻译和未折叠蛋白质应答[12]
真核细胞翻译起始源于形成异三聚体eIF2复合体。该复合体包括α-亚基,结合tRNA的β-亚基和结合GTP的γ-亚基。eIF2复合体负责在40S亚基上装载Met-tRNAi。结合mRNA后,43S复合体作为骨架,招募其他几个蛋白质,包括eIF3,到5′加帽的mRNA。随后,结合了帽子的真核翻译起始因子4F(eukaryotic translation initiation factor 4F,eIF4F)也添加到48S起始前复合体(pre-initiation complex),从5′到3′到扫描mRNA,定位翻译起始密码子(Translation initiation codon)。此后,60S核糖体亚基加入,蛋白质合成起始。结合到mRNA poly(A)尾部的多聚腺苷结合蛋白(Polyadenine-binding protein,PABP)也参与激发蛋白质合成。外界不同刺激可以分别激发哺乳动物4个激酶,磷酸化alpha亚基(eIF2α),失活eIF2复合体。这四个激酶是:eIF2α激酶4(eIF2α kinase 4,也称GCN2),血红素调控的抑制分子(Heme-regulated inhibitor,HRI),ER胁迫时被激活的PKR样内质网激酶(PKR-like endoplasmic reticulum kinase,PERK)和双链RNA激活的蛋白激酶(Double-stranded (ds) RNAactivated protein kinase,PKR)。
宿主细胞的蛋白质翻译机器既是病毒复制所需,也是宿主抗病毒应答的关键。正链RNA病毒需要限制宿主合成mRNAs,有利于病毒蛋白质合成。冠状病毒复制周期的多个阶段都与内质网结合。因此,冠状病毒感染时,可能导致内质网胁迫。表达冠状病毒的几个蛋白质如S蛋白,可以诱导内质网胁迫,与冠状病毒感染的细胞的表型一样。随后诱导未折叠蛋白质应答(Unfolded protein response,UPR),减轻PERK诱导的eIF2α磷酸化造成的翻译抑制,刺激蛋白质折叠,激发凋亡。冠状病毒控制UPR的机理远没HCV的研究透彻。冠状病毒感染可能以控制PERK活性为主来调控翻译水平。PKR是丝氨酸/苏氨酸蛋白激酶(Serine/threonine protein kinase),被RNA病毒感染的标志物dsRNA激活,是针对RNA病毒感染的先天免疫的重要分子,上调抗病毒基因表达,包括产生干扰素(Interferons,IFNs)。冠状病毒以各种方式抵消PKR介导的信号传导,防止因为eIF2α磷酸化而关闭翻译。鸡传染性支气管炎病毒(Infectious bronchitis virus,IBV)通过激活PKR,以及诱导生长阻断和DNA损伤诱导的34蛋白(Growth arrest and DNA-damage-inducible 34 protein,GADD34)弱拮抗PKR,降低IBV-感染细胞中eIF2α磷酸化。MHV感染时,持续eIF2α磷酸化和阻遏GADD34表达导致抑制细胞mRNA的翻译,有利于MHV感染。MERS-CoV ORF4a可能与病毒dsRNA结合,防止被PKR检测到,拮抗PKR-诱导的形成胁迫颗粒(Stress granule),防止翻译抑制。传染性肠胃炎病毒(Transmissible gastroenteritis virus,TGEV)调控宿主细胞翻译。其蛋白7与蛋白磷酸酶1相互作用,促进eIF2α去磷酸化。PP1是宿主抗病毒应答的关键调节因子。SARS-CoV和IBV的S蛋白与eIF3F相互作用,调控宿主翻译,包括后期促炎症细胞因子白细胞介素6和8的表达。这种相互作用可能在冠状病毒致病机理中发挥重要调控功能。
除调控eIF2α磷酸化,冠状病毒也通过其他方式调控宿主的翻译机器。alpha-和betacoronaviruses病毒的nsp1蛋白抑制翻译起始的多个步骤。SARSCoV nsp1抑制48S起始复合体形成,干扰其转变为80S起始复合体。SARS-CoV nsp1多功能,可以直接结合40S核糖体亚基并抑制其在翻译中的功能。nsp1-40S亚基复合体诱导切割宿主细胞mRNA如IFN-β mRNA,更大范围地阻遏细胞翻译,但是也有报道诱导大量IFN-β。MERS-CoV nsp1选择性抑制细胞核产生的mRNAs的翻译,但是不影响细胞浆中产生的病毒mRNAs的翻译。MERS-CoV nsp1不与40S核糖体亚基结合。病毒的翻译调节因子(translation modulator)与致病有关。
宿主环孢亲和素(Cyclophilin):环孢菌素A(Cyclosporin A,CsA)是免疫抑制剂,结合细胞环孢亲和素(cyclophilins,Cyps),形成Cyp-CsA复合体,抑制钙调磷酸酶活性。其阻止细胞核因子活化的T细胞(nuclear factor of activated T cells,NF-AT)去磷酸化,从细胞浆转位到细胞核,阻止免疫基因如IL-2的转录。已经鉴定的17个Cyps中,9个是CsA靶标。Cyps也是肽酰基脯氨酰基异构酶(peptidylprolyl isomerases,PPIases),许多还具有分子伴侣和折叠酶活性,有利于蛋白质折叠。Cyps参与各种信号途径,凋亡、RNA剪切。环孢亲和素,尤其是细胞浆CypA和结合内质网的CypB,是许多RNA病毒复制周期组分,也是必需的宿主组分。(1) Cyps是重塑细胞膜为HCV复制细胞器必需;(2) CypA帮助HCV多聚蛋白加工;(3) 低水平CypA稳定HIV-1衣壳,确保进入细胞核,以免病毒粒子在细胞浆中变得不稳定。Alisporivir(DEB-025;Debio-025) 是环孢亲和素CsA类似物,具有高效的抗丙型肝炎病毒(HCV)活性,但没有母核的免疫抑制活性。这说明环孢亲和素在HCV复制中的作用,以及成靶性。微摩尔浓度CsA或Alisporivir可以抑制很多冠状病毒如SARS-CoV、HCoV-229E、MHV、HCoVNL63和FCoV。线粒体CypD是线粒体通透性孔(permeabilization transition pore)的一部分,参与猪流行性腹泻病毒(porcine epidemic diarrhea virus,PEDV)和HCoV-NL63诱导的半胱天冬酰胺酶非依赖性凋亡,CsA可以抑制该过程。质谱和酵母双杂交都证实CypA与SARS-CoV N 蛋白相互作用。酵母双杂交发现多个Cyps(CypA、CypB、CypH和CypG)及相关PPIases(FK506-binding protein 1A和1B)可能结合SARS-CoV nsp1。Caco-2或Huh7细胞中,shRNA-介导敲除CypA,表达降低到正常水平的3%,几乎可以完全阻断HCoV-NL63复制,降低HCoV-229E 复制>1log。影响CypA稳定性和功能的单核苷酸多态性(single-nucleotide polymorphisms)可以降低不同HCoV-229E复制。Cyps主要作用在NF-AT信号途径。Cyp抑制剂可以作为工具药物,探索宿主因子在冠状病毒复制中的作用,但是作为宿主靶向治疗(host-directed therapeutics)冠状病毒感染尚缺乏证据。
冠状病毒RNAs 5′-和3′-近端区域含有其RNA合成的关键调控元件。RNA-结合蛋白常可以作为冠状病毒基因组复制的增强子。冠状病毒基因组两端折叠为高阶RNA结构,稳定该分子,参与分子内部和分子间相互作用,有利于病毒复制。病毒和宿主的蛋白质结合这些结构,驱动或者调控翻译、复制和亚基因组RNA合成(表2)。
宿主细胞蛋白polypyrimidine tract-binding protein(PTB;or heterogeneous ribonucleoprotein protein(hnRNP) I)结合TGEV和MHV基因组的5′前导序列(leader sequence)。PTB结合MHV 5′-五核苷酸UCUAA重复序列UCUAA,是RNA合成的关键。HnRNP Q或SYNCRIP也结合MHV基因组的5′-端,如果敲除,MHV RNA合成和病毒复制减少。锌指 CCHC-型和RNA-结合模体(zinc finger CCHCtype and RNA- binding motif 1,MADP1)结合SARSCoV和IBV基因组的5′端。MAPD1沉默也干扰病毒RNA合成,减少IBV复制。除了结合冠状病毒RNA 5′- UTR(5′-untranslated region)的蛋白质。5′-UTR约310 nucleotides(nts),含前导序列(leader sequence)和转录调控序列(transcription regulatory sequence,TRS),具有巢病毒目都保守的核心TRS模体(core-TRS motif)5′-CUAAAC-3′。也有蛋白质结合3′UTR和/或poly(A)-尾。比如线粒体顺乌头酸酶(mitochondrial aconitase),被heat-shock protein (hsp)40、hsp60和mitochondrial hsp70复合体所稳定,结合MHV 3′ UTR 位于poly(A)尾上游3′-端42核苷酸。P100辅助激活因子(p100 coactivator)结合TGEV′UTR和/或poly(A) 尾。PABPs与TGEV、BCoV和IBV基因组相互作用,促进其有效复制。PABPs结合病毒RNA确保有效翻译和mRNA稳定。RNA-亲和纯化和质谱发现几个hnRNPs结合TGEV基因组3′-端约500核苷酸。RNA结合蛋白可能在复制酶基因翻译时,调控冠状病毒ORF1a/1b移码。annexin A2结合IBV滑动序列(slippery sequence)。该滑动序列是核糖体停滞和参与ORF1b表达所需。宿主蛋白也可能间接影响病毒RTC活性。
2.5 冠状病毒复制需要的宿主因子的系统生物学研究[13]
包括转录组学、代谢组学、蛋白质组学和功能基因组学等在内的系统生物学方法将感染系统作为整体考虑,优点是无偏好,可以鉴定冠状病毒复制需要的蛋白质、细胞途径。芯片研究SARS-CoV-感染的外周血单核细胞(peripheral blood mononuclear cell),上调的细胞因子有IL-8、IL-17,激活巨噬细胞和凝血途径。肺部组织尸检也提供了SARS-CoV感染的病理,尤其是炎症和细胞因子应答。MHV-JHM感染培养的中枢神经系统细胞中,126个基因差异表达。其中大多数是调控胞内先天免疫如NF-κB信号通路和IFN信号传导。MHV-A59-感染的L细胞中,趋化因子、RNA和蛋白质代谢、凋亡等途径差异表达。芯片分析发现MHV感染的LR7细胞下调包括蛋白质翻译相关许多基因的转录,提示感染时,因为胁迫应答和mRNA降解(mRNA decay),宿主关闭了蛋白质翻译系统。mRNA水平和蛋白质丰度未必直接相关。转录水平的结果还需要用其他实验,如细胞蛋白质水平进行验证。
相互作用组酵母双杂交发现冠状病毒RNA聚合酶复合体的亚基BTF3和ATF5、细胞色素氧化酶II(NADH 4L)与SARS-CoV nsp10相互作用。当然,这个结果还需要在SARS-CoV-感染细胞中进行验证。SARS-CoV helicase (nsp13)与DDX5相互作用,DDX1与SARS-CoV和IBV nsp14相互作用,促进IBV复制。DDX1调控sg mRNA合成。该过程受GSK-3-介导的N 蛋白磷酸化调控。分析14 个SARS-CoV ORFs 及片段与宿主相互作用发现:nsp1与包括CypA在内的免疫亲和素家族相互作用。细胞E3泛素连接酶RCHY1与SARS-CoV nsp3 SUD(SARS unique domain)功能域相互作用,可能参与下调抗病毒因子p53。SUD蛋白能够特异结合宿主mRNA 3′-不译区广泛具有的GGGG序列,提示SUD可能参与调控宿主mRNA代谢[14-18]。
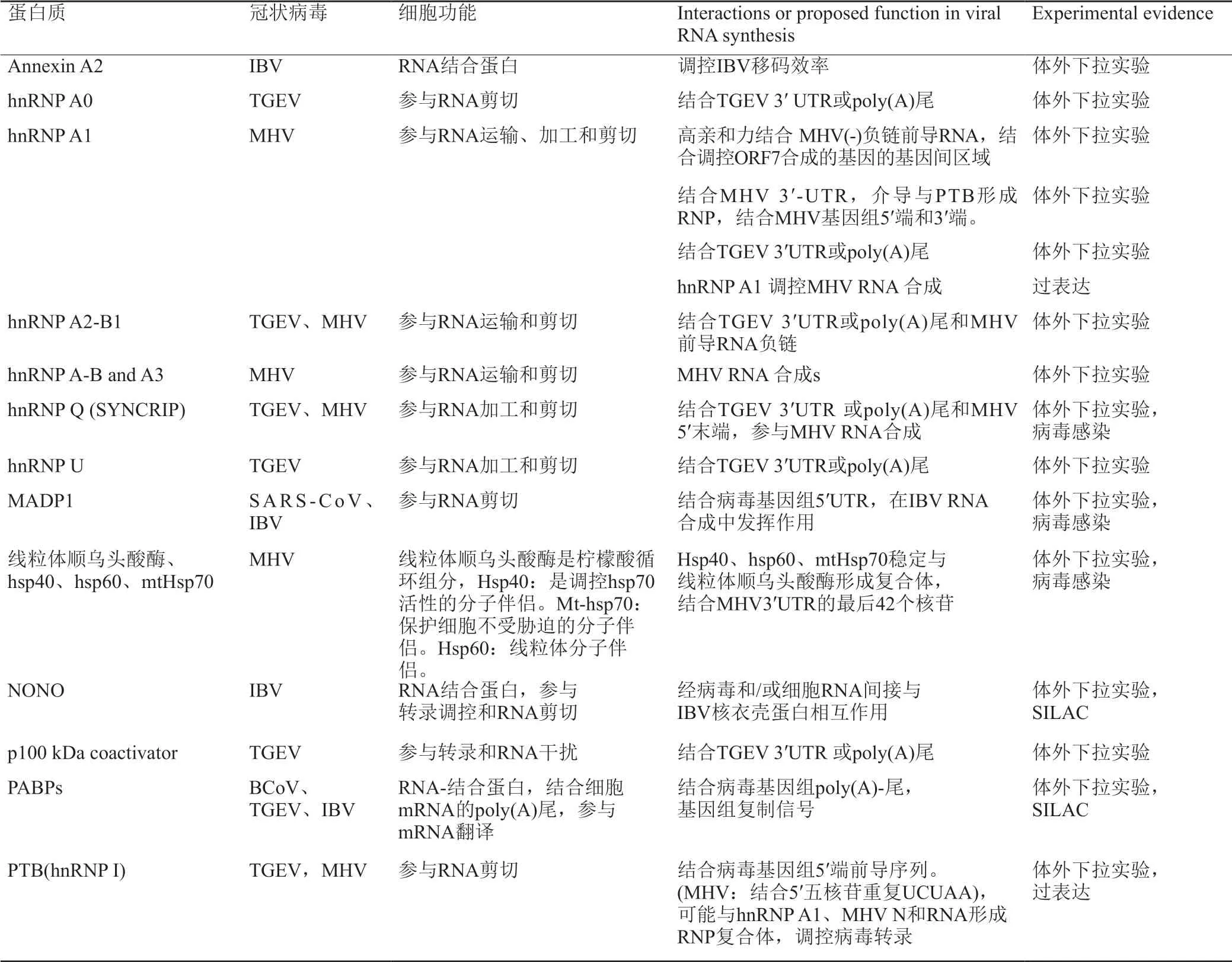
表2 与不同冠状病毒RNA相互作用的RNA结合蛋白
稳定同位素标记的氨基酸细胞培养(Stable isotope labeling with amino acids in cell culture,SILAC)是一种基于质谱的蛋白质组学技术,可以确定两种实验条件下样品中蛋白质丰度,比如感染和未感染细胞。SILAC研究发现IBV感染上调NF-κB和AP-1依赖性途径。SILAC结合pull-down和质谱技术,研究表达IBV N蛋白的293T相互作用组,得到142个细胞蛋白质。其中许多蛋白质与RNA相互作用,如核糖体和核仁蛋白质,螺旋酶,以及hnRNP。
基于SILAC的定量蛋白质组学(SILAC-based quantitative proteomics)比较表达SARS-CoV复制子的BHK-21细胞,找到宿主蛋白质43个上调,31个下调。许多细胞过程的多功能调控因子BAG3也被上调。敲除实验证明,SARS-CoV和其他病毒有效复制需要BAG3。下调的有参与蛋白质翻译和信号传递的蛋白质Cdc42和RhoA。基于SILAC的定量蛋白质组学研究高尔基体富集的亚细胞组分发现MHV感染耗尽几个分泌途径的蛋白质,富集核糖体蛋白质。SiRNA-介导敲降其中3个蛋白质:C11orf59、GLG1和sec22b后,感染性子代病毒的复制或释放增加,过量表达这些蛋白质则具有相反效应。宿主细胞分泌途径对于冠状病毒复制很关键。蛋白质组分析纯化的SARS-CoV病毒粒子发现,除nsp2(各冠状病毒中变异较大),nsp3(各冠状病毒中变异较大)和nsp5等病毒蛋白外,病毒粒子还结合有172个宿主蛋白质。其中有几个是COPI途径的蛋白质,与病毒粒子形成途径(ERGIC)吻合。SARS-CoV nsp2可与宿主蛋白抑制素1(prohibitin 1,PHB1)和抑制素2(prohibitin 2,PHB2)相互作用。在病毒感染过程中,可能破坏宿主细胞信号通路。
系统遗传学方法:利用一组杂交小鼠,筛选影响SARS-CoV感染结局的宿主位点发现E3泛素连接酶Trim55是重要决定因子。Trim55在血管套和炎症中发挥重要作用。MERS患者免疫和炎症应答相关的遗传背景与病毒毒株共同决定了感染结局。比较不同MERS-CoV分离株对宿主细胞的转录组影响发现,病毒PLpro和ORF4a参与免疫逃避,影响宿主STAT3途径,对肺部炎症和细胞修复有影响。这些效应主要出现在感染后期。
蛋白激酶是信号传导的关键调控因子,控制许多细胞过程,在许多正链RNA病毒的复制周期中发挥作用。激酶组siRNA(kinome-wide siRNA)筛选发现许多宿主细胞激酶影响SARS-CoV复制,其中包括40个有效促进病毒复制的"proviral"蛋白质。如复杂脂类代谢和早期分泌途径(COPI-coated vesicles)。抗病毒效应的蛋白质包括PKR和CDK6。相比其他病毒,更多蛋白质参与SARS-CoV复制,这也说明宿主细胞对SARS-CoV复制的影响大。p38 MAPK途径调控IL-6,IL-8和IL-10介导的促炎症细胞因子信号传递。网格蛋白介导的内吞和运输到溶酶体对于MHV融合、进入很关键,溶酶体蛋白酶是该过程所需条件。综合利用CRISPR/Cas9-介导的基因组编辑(genome editing)或单倍体遗传筛选(haploid genetic screen)等技术,与系统生物学技术结合,可以鉴定更多冠状病毒复制所需的宿主细胞因子。
2.6 冠状病毒诱导宿主细胞周期失控
负责细胞分裂的细胞周期被严格调控,分为G1,S,G2和M。细胞存活、防止不受控制的细胞分裂都需要调控细胞周期。周期蛋白依赖的蛋白激酶(cyclin-dependent protein kinases,CDKs)有序控制细胞周期。细胞周期进行需要周期蛋白调控亚基激活不同CDK。进入DNA复制,CDK/cyclin复合体磷酸化,激活或失活其靶蛋白,协调进入下一轮周期。类似其他病毒,冠状病毒大量调控、阻遏细胞周期进行,受益于被阻断的生理状态。IBV-感染细胞阻遏在S期,激活细胞DNA损伤应答,对病毒复制有利。因为S期上调DNA复制所需蛋白质,病毒可以招募到细胞浆。例如与冠状病毒nsp14相互作用的细胞DExD/H家族的RNA螺旋酶(RNA helicase)DDX1被冠状病毒劫持,用来增强其复制。SARS-CoV nsp14近C端区域有鸟嘌呤-N7-甲基转移酶活性,在RNA加帽过程中起重要作用,其活性依赖nsp14近N端核酸外切酶活性区域。DDX1也与IBV N蛋白相互作用,与MHV-JHM N蛋白的磷酸化形成复合体,有利于平衡合成sg mRNA和基因组RNA。冠状病毒nsp15相互作用并抑制抑癌蛋白视网膜母细胞瘤蛋白(retinoblastoma protein,pRb),平时被pRb阻遏的蛋白质的表达增加,进入S期的细胞增多。类似效应在MHV感染细胞中也存在,G1期进入S期所需要的的pRb低度磷酸化被降低。过量表达SARS-CoV 3a蛋白导致G1阻断,抑制细胞增殖。
MHV感染下调cyclin-dependent kinase 6(CDK6) ,在病毒诱导的促进病毒复制的细胞周期阻遏在G0/G1期发挥作用。SARS-CoV类似效应,过量表达N 蛋白,降低CDK4CDK6激酶活性,限制细胞周期推进。CDK6激酶参与细胞周期从G1推进到S期,耗尽CDK6导致细胞周期阻断在G1期。
部分冠状病毒诱导p53,将细胞周期阻断在S或G2/M期。TGEV N 蛋白激活p53,积累细胞周期相关激酶如cdc-2和cyclin B1。同步在S或G2/M期的细胞有利于TGEV RNA和病毒产生,以及IBV复制。MHV nsp1也具有类似机制。nsp1激活p53,提高p21水平,降低CDK2-cyclin E水平。pRb滴度磷酸化和G1细胞周期阻断。SARS-CoV感染导致减少p53表达水平。为了抵消p53的抑制效应,SARS-CoV nsp3(分子量>200kDa,具有poly(ADP-ribose)结合功能域,蛋白酶、去泛素酶和de-ISGylase功能域)稳定E3连接酶RCHY1,降低其表达。RCHY1介导p53泛素化并降解。但是,调控细胞周期、病毒复制和/或致病的具体机理仍然不清楚。缺乏p53表达的细胞,SARS-CoV复制被明显增强。SARS-CoV或HCoV-NL63 PL2pro降解p53,抵消p53的作用。PL2pro去泛素化,促进降解p53,降低p53-介导的抗病毒免疫应答。
2.7 宿主对冠状病毒的先天免疫应答以及病毒的对抗策略
宿主先天免疫试图遏制病毒复制。病毒采用多种策略逃避宿主免疫应答。宿主细胞浆中正链RNA病毒复制中间体dsRNA 和5′-三磷酸RNA分子可以激发免疫。几乎所有细胞的细胞浆中都存在Rig-Ilike receptor(RLR)家族感受器如retinoic acid-inducible gene 1(RIG-I)、病毒感染时合成2′,5′-寡腺苷酸的2′,5′-寡腺苷酸合成酶(2′,5′-oligoadenylate(2~5A) synthetases,OAS)和黑色素瘤分化相关蛋白质5 (melanoma differentiation-associated protein 5,MDA-5)负责识别dsRNA和5′-三磷酸RNA分子。2~5A结合和激活RNase L。RNase L是宿主核糖核酸酶,负责全局降解宿主和病毒的RNA。细胞表面的TLR或者免疫细胞内吞体中的TLR如TLR3、TLR4也可以识别病毒核酸或者蛋白质。SARS-CoV M 蛋白被TLRlike途径识别,与标准的TRAF3-介导的信号途径独立,随后激活转录因子IRF3,IRF7(IFN-regulatory factor 3 and 7)和NF-κB,表达并分泌I型IFN和促炎症细胞因子,后者反馈激活JAK-STAT信号通路,诱导表达诸多具有抗病毒功能的干扰素刺激基因(interferon-stimulated genes,ISGs),被感染细胞进入抗病毒状态。ISGs靶向病毒复制周期的几乎所有步骤,限制病毒复制。p38丝裂原活化的蛋白激酶(p38 mitogen-activated protein kinases,MAPKs)参与诱导炎症细胞因子IL-6和IL-8,参与抗病毒感染。IBV诱导p38 MAPK的负调控因子-双特异性磷酸酶1(dualspecificity phosphatase 1,DUSP1),拮抗IL-6和IL-8表达。尚不知晓IBV病毒具体哪个(些)分子导致上述效应。整个信号通路的多个节点如RIG-I、TANK结合激酶(TANK-binding kinase 1,TBK1)和TNF受体相关因子(TNF receptor-associated factor 3,TRAF3)都可能受到磷酸化、泛素化等翻译后修饰的调控,如Lys63-泛素化。
先天免疫涉及包括细胞因子、趋化因子、补体、巨噬细胞、树突状细胞等组成的复杂网络,旨在阻止病毒进入和繁殖。但病毒也共进化出逃避/阻断宿主先天免疫的组分。这些组分用来避开宿主免疫监视,操纵宿主先天免疫应答。
冠状病毒感染中,先天免疫的作用体现在至少3个方面:重症SARS患者伴随先天免疫信号传递异常或者过度活化,肺部产生更多干扰素和促炎症细胞因子如L-1、IL-6、IL-8、CXCL-10和TNF-α。不同MERS-CoV 分离株(MERS-CoV Eng 1 vs.SA 1) 感染人培养细胞的转录组发现,病毒序列差异和先天免疫逃避有关,导致的免疫应答不同。随后,STAT3差异激活导致IFN、NF-κB和IRF的活化或者抑制。冠状病毒利用复杂机制使病毒复制机器(viral replication machinery)避开宿主细胞浆中先天免疫感受器的监视。除复制细胞器对病毒PAMPs的保护外,冠状病毒也主动进攻或者躲避宿主的先天免疫应答。
病毒采用多种因子、多种途径压制宿主的先天免疫应答。除抑制细胞mRNA翻译外,SARS-CoV nsp1降低被感染细胞中磷酸化STAT1(phosphorylated STAT1,p-STAT1)的量,以及IRF3二聚化,阻断产生IFN相关的信号传递,影响细胞周期,但不诱导凋亡。其他alpha-和betacoronaviruses的nsp1蛋白(相对变化大)也抑制I型IFN信号传递,大多数冠状病毒复制酶多聚蛋白N端亚基具有关闭宿主免疫应答的活性。Nsp1缺陷病毒对IFN更敏感,这也提示nsp1是IFN拮抗剂。但Nsp1拮抗先天免疫、选择性降解宿主mRNA则还需要证据。生化证据表明,pp1a多聚蛋白更下游,许多冠状病毒的papain-like protease 2 domain(PL2pro)和TGEV nsp3的PL1pro功能域,具有去泛素化酶(deubiquitination,DUB)活性。DUB活性可以除去先天免疫信号传递因子的泛素,阻止抗病毒状态的产生,生化实验证明SARS-CoV和MERSCoV PL2pro过量表达可以降低干扰素信号传递。MHV PL2pro去泛素化并结合TBK1,以及IRF3。
除了保守的复制酶,冠状病毒其他辅助蛋白也可以压制宿主先天免疫。这些蛋白质可能是不同病毒株系为此而从不同来源获得的。细胞培养中,这些辅助蛋白并非必需。MHV ORF2a编码的磷酸二酯酶(phosphodiesterase)ns2拮抗宿主RNAse L激活。这对于在天然宿主中复制非常重要。SARSCoV ORF3b和ORF6阻断干扰素产生和信号传递。MERS-CoV ORFs 4a,4b和5也阻断干扰素信号传递。SARS-CoV E蛋白是病毒编码的离子通道-病毒孔蛋白(viroporin),具有促进NLRP3炎症体活性,影响炎症进程,导致大量产生IL-1β,导致宿主免疫病理性变化。
ORF6是病毒-宿主相互作用的关键,调控胞内环境。调控感染过程中的进入细胞核的动力学(图4),病毒可以控制先天免疫、适应性免疫、凋亡和细胞胁迫应答网络。ORF6是63aa的ER/Golgi膜蛋白,C-端尾部面向细胞浆,N-端位于ER腔或者ER膜。用IFN-β或IFN-γ处理,ORF6能够阻断STAT1/STAT2/IRF9和STAT1/STAT1复合体进入细胞核。ORF6减少可以抑制STAT1依赖的基因表达,这需要C-端10aa。ORF6招募细胞核进入因子到ER/Golgi膜。KPNA2特异性结合ORF6的C-端,将KPNA2滞留在ER/Golgi膜上,随后,招募KPNB1到膜复合体,限制STAT1复合体进入细胞核必需的KPNB1的可利用度,结果是ORF6阻止STAT1复合体进入细胞核。KPNB1是经典的进入细胞核途径的共同组分。ORF6因此可以影响其他信号途径。ORF6阻断含有经典入核信号的蛋白,但是对于不含这些经典入核信号的蛋白则无效。ORF6可以使一般无毒的MHV-A59毒性增加。MHV/ORF6病毒数量比野生型病毒多。MHV的辅助蛋白可能与逃避免疫监视有关。细胞核转运蛋白的水平随细胞类型、年龄而不同,其可利用度与感染密切程度相关。
免疫应答是保护还是致病?宿主免疫系统涉及多个组织、细胞和分子,通过有效识别和去除致病菌,保护宿主应对感染。SARS-CoV感染严重程度与病毒特异性免疫应答相关,抑制肺泡巨噬细胞和不能有效激活树突状细胞,可以延缓病毒特异性免疫应答,加剧疾病。如果I型干扰素应答和炎症性单核细胞-巨噬细胞应答失控,SARS-CoV感染小鼠产生致死性肺炎。如果抑制NF-κB介导的炎症,SARS-CoV感染小鼠存活率则提高。IAV破坏宿主组织与过度激活NLRP3(NACHT,LRR and PYD domains-containing protein 3),HMGB-1(highmobility group box 1 protein),IL-1β有关。TNFSF10,HDAC4(histone deacetylase 4),HDAC5表达与TNFα,NF-κB,cox-2(cyclooxygenase 2)水平负相关,其表达增加与预后良好相关。
2.8 细胞因子风暴和免疫病理的关系
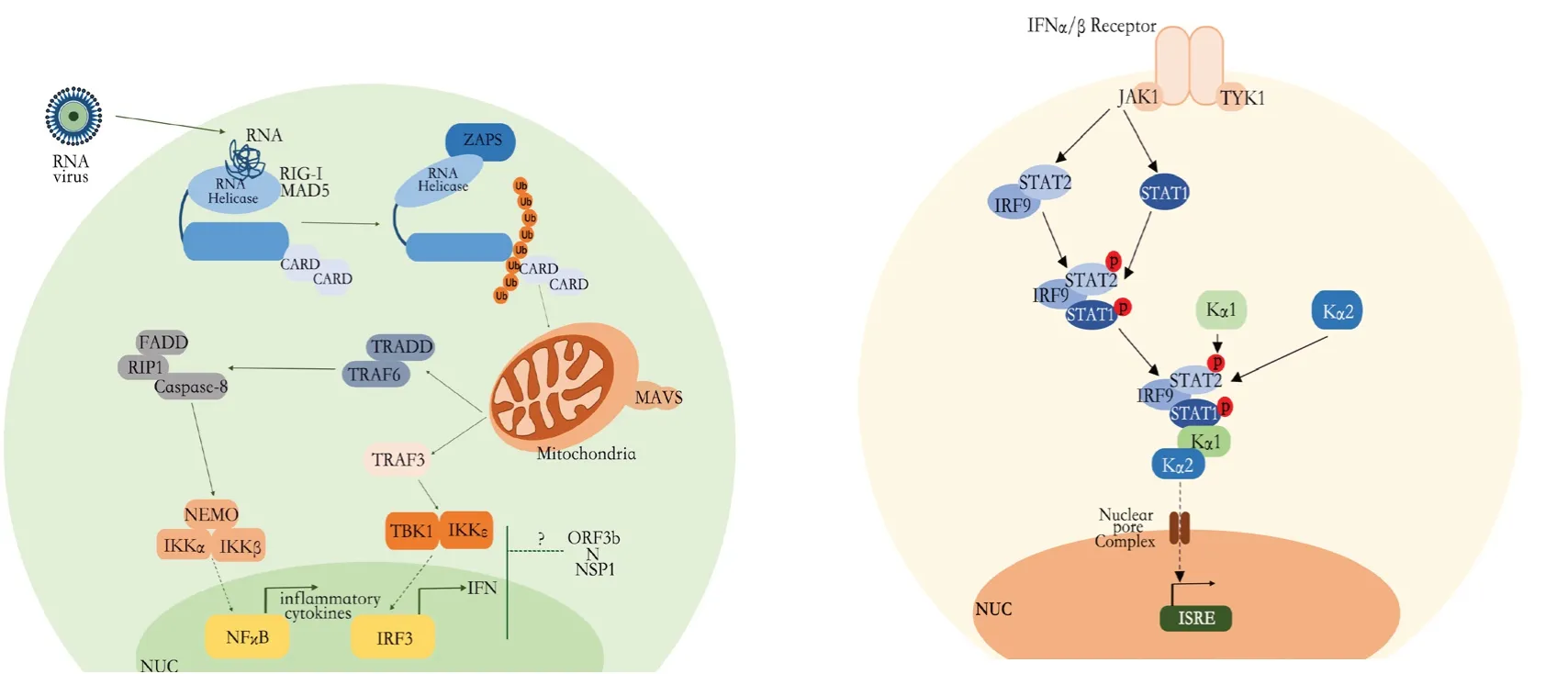
图4 产干扰素细胞(左)和含干扰素受体细胞(右)的信号通路。
如细胞因子和趋化因子应答迅速且协调,则可以作为先天免疫一线防御病毒感染。如果失控或者过量产生,则导致病理变化,称为细胞因子风暴(cytokine storm)[19]。目前尚无SARS和MERS感染过程中,促炎症细胞因子和趋化因子参与肺部病理的直接证据。患者的相关性证据提示炎症应答增加,与冠状病毒致病有关。
SARS-CoV感染过程中的细胞因子和趋化因子应答,SARS-CoV主要大量感染呼吸道和肺泡上皮细胞,一般不易成功感染血液细胞如树突状细胞、单核细胞-巨噬细胞和其他PBMC衍生的细胞。SARSCoV感染后DCs产生低水平的抗病毒细胞因子IFNαβ,中等程度上调促炎症细胞因子TNF和IL-6,明显上调炎症趋化因子CCL3、CCL5、CCL2和CXCL10。SARS-CoV-感染的巨噬细胞IFN和其他促炎症细胞因子的产生延迟但增加。SARS-CoV-感染的呼吸道上皮细胞(airway epithelial cells,AECs)也大量产生CCL3、CCL5、CCL2和CXCL10。延迟但大量产生这些细胞因子和趋化因子导致针对SARS-CoV的先天免疫失控。与无并发症的SARS患者比较,SARS重症患者血清促炎症细胞因子(IFN-γ、IL-1、IL-6、IL-12和TGFβ)和趋化因子(CCL2、CXCL10、CXCL9和IL-8)水平高。重症SARS患者的抗炎症细胞因子如IL-10水平很低。与健康人和中等程度患者比较,除了促炎症细胞因子和趋化因子,致死性SARS患者水平增加的有IFN(IFN-α和IFN-γ)和IFN刺激基因(IFN-stimulated genes,ISGs)(CXCL10和CCL-2)。IFNs和ISGs可能参与SARS免疫病理。SARSCoV-感染的AECs,DCs和巨噬细胞的细胞因子、趋化因子应答失控或者增加,可能具有病理作用。SARS感染者血清中促炎细胞因子(IL-1β、IL-6、IL-12、IFN-γ、IP-10和MCP-1)的增加与SARS感染者肺部炎症和广泛的肺损伤有关。年龄影响感染结局,适应性免疫随着年龄增加而降低。但是,先天免疫还需要研究,尤其是年轻人和老年人的病毒易感性相关SNP。SARS-CoV康复者才增加IFN-γ、IL-4、IL-10。SARS-CoV患者大幅上调IL-6。IL-6,IL-8和MCP-1(monocyte chemoattractant protein 1)和IP-10与疾病严重程度和死亡相关。芯片定量研究外周血单核细胞的细胞因子表达发现:所有感染者都提高促炎症细胞因子表达,但是几乎未见IFN-α或β表达增加。这可能是后两者在被感染细胞中稳定,或者IFN拮抗差异(SARS-CoV编码大量诱导IFN的dsRNA,但是未见IFN产生)。SARS-CoV感染如何诱导细胞因子?与病毒致病机理有何相关?与疾病严重程度相关的标记分子目前包括人白细胞抗原I(human leukocyte antigen class I,HLA-I) (B*0703),HLA- II (DRB1*0301),甘露糖结合蛋白(mannose binding protein),OAS1,MxA。但是,样本小且不容易获得,所以较难重复。IL-12RB1,IFN-γ,rantes等多态性与SARS疾病严重程度相关。这些现象的机理,目前尚不清楚。可能有以下几个方面:病毒编码的蛋白直接拮抗IFN产生途径,而且,病毒可能编码多个干扰素拮抗分子。病毒可能在某个隐秘的部位复制,宿主无法检测其RNA存在,比如双层膜结构中。MHV或者SARS-CoV干扰的细胞,确实不产生IFN-β。但是在病毒感染后,加入Sendai病毒或者poly(I:C),细胞产生大量IFN-β。MHV感染的DC细胞产生很低水平I型IFN。SARS-CoV感染的Caco-2和HEK293细胞,几乎不产IFN-α,IFN-β,IFN-λ。Caco-2细胞而不是HEK293细胞诱导IP-10和IL-8。但是,产生差异的原因未知。这可能对解释人和小鼠差异有关。冠状病毒抑制的途径可以被其他诱导因素激活,细胞培养模型中,IFN信号传递途径未被完全抑制。连续研究病毒RNA的定位、复制动力学和翻译,有助于理解该现象。
MERS-CoV感染中的细胞因子和趋化因子,类似SARS,MERS-CoV也感染人呼吸道上皮细胞,诱导大量但延迟的IFN和促炎症细胞因子(IL-1β,IL-6和IL-8)。MERS-CoV可以在未激活和激活的人单核细胞-巨噬细胞、DCs中存活,但MERS-CoV只在激活的T细胞中复制。这点不同于SARS-CoV。SARSCoV不能有效感染单核细胞-巨噬细胞、DCs和T细胞。MERS-CoV感染单核细胞来源的细胞株THP-1,人外周血单核细胞衍生的巨噬细胞(peripheral blood monocyte-derived macrophages)和DC,诱导迟缓但高水平的促炎症细胞因子和趋化因子如CCL-2、CCL-3、CCL-5、IL-2和IL-8。但是,除了浆细胞样树突状细胞(plasmacytoid dendritic cells,pDC)在感染时产生大量IFN,单核细胞-巨噬细胞和DC都不大量产生IFN-α/β。与轻症比较,重症MERS患者的血清促炎症细胞因子(IL-6、IFN-γ、TNF-α、IL-17、IL-15和IFN-α)和趋化因子(IL-8,CXCL10和CCL5)高。与其正相关的是肺部和外周血中嗜中性粒细胞和单核细胞数量增加。这提示这些细胞可能参与肺部病理。
2019-nCov和MERS,SARS都属于冠状病毒,但是其细胞因子风暴特点不同。初期:IL1B、IL1RA、IL7、IL8、IL9、IL10、碱性FGF、GCSF、 GMCSF、IFNγ、IP10、MCP-1、MIP1A、MIP1B、PDGF、TNFα和VEGF均升高。IL5、IL12p70、IL15、Eotaxin和RANTES无改变。ICU(重症)和非-ICU病人(轻症)比较:IL2、IL7、IL10、GCSF、IP10、MCP-1、MIP1A和TNFα升高。猜测的机制为:IL1B、IFNγ、IP10、MCP-1激活Th1。GCSF、IP10、MCP-1、MIP1A和TNFα与疾病严重程度相关。与SARS-Cov不同,抑制免疫的Th2细胞因子IL-4,IL-10在2019-nCov也升高。
细胞因子风暴与冠状病毒感染大量炎症的原因,以及驱动肺部病理损伤的宿主因素总体不清楚。但是,在炎症起始和进展过程中发挥作用的因素正被逐步阐明。(1)病毒快速、大量复制:体内外感染的情形都是SARS-CoV和MERS-CoV很快大量复制,细胞毒性效应增强,被感染的上皮细胞产生大量促炎症细胞因子和趋化因子。后者反馈协调大量炎症细胞浸润肺部。SARS-CoV和MERSCoV病毒数量与疾病严重程度相关。(2)hCoV感染呼吸道和/或肺泡上皮细胞:来自小鼠等动物模型的研究都提示病毒主要感染呼吸道上皮细胞,以及肺泡上皮细胞(I型和II型肺细胞pneumocyte)。(3)IFN应答延迟: SARS-CoV和MERS-CoV编码大量结构蛋白和非结构蛋白,拮抗IFN应答。感染不久,hCoV就大量复制,产生大量抑制IFN应答的蛋白质,提示IFN应答早期拮抗可能滞后或者逃避先天免疫应答。延迟的IFN 信号传递进一步协调IMM应答,致敏T细胞凋亡,炎症应答失控。(4)单核细胞-巨噬细胞和嗜中性粒细胞积累:冠状病毒感染后,人体肺部积累这些炎症细胞。与疾病致死性相关的细胞因子和趋化因子主要来自这些细胞。病毒复制迅速、大量炎症细胞浸润、促炎症细胞因子/趋化因子增多,导致急性肺损伤(acute lung injury,ALI)和急性呼吸窘迫症(acute respiratory distress syndrome,ARDS)。免疫应答失控导致肺部免疫病理,有害临床症状。
3 冠状病毒防控措施研发
抗冠状病毒药物研发面临的挑战既有RNA病毒共性问题,也有冠状病毒特有问题。抗病毒靶标有蛋白酶、解旋酶和聚合酶抑制剂。主要靶标是病毒感染必需的宿主细胞机器,或者干扰病毒致病必需的宿主先天免疫应答。新药研发比较漫长,为了提高治愈率,降低死亡率,老药新用是省时、成本效益好的开发高效抗病毒药物方法。冠状病毒感染的治疗方法包括康复者血清(convalescent sera)、皮质激素(corticosteroids),抗病毒药物(antiviral therapeutics)如干扰素(interferons)、广谱抗病毒的核苷类似物如利巴韦林(ribavirin)、蛋白酶抑制剂(protease inhibitor),见表3、图5。
3.1 治疗方法
康复者血清:将MERS-CoV免疫过的骆驼的血清输给感染小鼠,小鼠肺部病理减轻、体重降低也变缓。康复者的MERS-CoV-特异性抗体滴度变化幅度大,不容易获得大量血浆供体,相关的临床研究后来撤销(NCT02190799)。因此,目前缺乏安全性和有效性的证据。
疫苗:针对冠状病毒的疫苗研发目标是诱导广谱抗体。这需要精准界定病毒保守的保护性抗原表位。宿主差异的T细胞应答特征,以及可能基于DC的疫苗设计。目前研究发现靶向嗜中性粒细胞和Th1,帮助建立组织驻留记忆(tissue-resident memory,TRM)和对不同亚型病毒具有交叉保护作用的免疫(heterosubtypic immunity)对于疫苗研发非常重要。通过单细胞转录组等确定外周的组织驻留细胞亚群及其功能非常关键。综合利用测序、晶体结构,分子进化,预测病毒演化,选择动力学,宿主内动态,基于测序的流行病学、基因组分化与适应,建立预测模型,现场实验数据操作流程标准化,数据归一化,全面分析免疫各方面很必要。
订书肽(Stapled Peptide):蛋白-蛋白相互作用(PPI)在许多生物过程中发挥重要作用,例如病毒的自组装。订书肽可能抑制病毒与宿主相互作用,或者病毒不同蛋白之间相互作用。具有α螺旋结构和富含正电荷的多肽可以穿过细胞膜。2000年,Verdine等发展了一种用碳碳键作为支架来稳定多肽α螺旋结构的方法。由该方法得到的多肽称为订书肽( stapled peptides) 。订书肽有更高的α螺旋程度,与靶标的结合能力增加5~5000倍。此外,订书肽能透过细胞膜,难被蛋白酶水解,在生物体内的半衰期较长。Aileron Therapeutics的两种订书肽ALRN-5281和ATSP-7041有一定功能。
氯喹(Chloroquine)具有直接的抗病毒作用,抑制包括黄病毒(flaviviruses),逆转录病毒(retroviruses)和冠状病毒(coronaviruses)在内的几种病毒的依赖pH的复制步骤。氯喹抗病毒作用的主要机制可能是抑制糖基化。氯喹可能与人细胞内糖修饰酶或糖基转移酶特异性相互作用。氯喹从1944年开始用来治疗疟疾,以后用途逐渐扩大。1951年,用于治疗类风湿关节炎。磷酸氯喹及其他4-氨基喹啉类抗疟药(如哌喹,氨酚喹等)可能干扰了疟原虫裂殖体DNA的复制与转录过程或阻碍了其内吞作用,从而使虫体由于缺乏氨基酸而死亡,主要对疟原虫的红内期起作用。
核苷(酸)类似物抑制剂(nucleotide and nucleoside analogue inhibitor,NI)是杂环或者糖成分被修饰的嘌呤或者嘧啶的类似物,是病毒聚合酶(Polymerase)的竞争性抑制剂(Competitive Inhibitor),也是最大、最重要的一类抗病毒药物。携带一个磷酸基团,代谢后就是核苷酸的NI前体药物称为核苷酸类似物(Nucleotide Analogue)。不含磷酸的NI药物称为核苷类似物(Nucleoside Analogue)。核酸合成沿着核酸的5'-3'进行。作为原料的核苷酸,首先被三磷酸化(Triphosphorylation),变成三磷酸核苷(NTP)。除了ATP,其它的NTP都是细胞中的激酶(kinase)合成的。进入细胞后,宿主或者病毒的激酶将这些前体或者前药代谢为活性三磷酸。RNA病毒复制所需的RNA复制过程在人体细胞中不会发生。病毒复制必须用自己的聚合酶,如RNA病毒的RNA复制酶(RdRp)。RNA病毒维持需要不间断地复制,因为RNA在宿主细胞中会被不断分解。而核苷(酸)类似物通过选择性的抑制病毒的聚合酶、错配或者替代,破坏RNA合成、结构、RNA-蛋白质相互作用或者蛋白质功能,阻止RNA病毒的复制。突变积累,病毒不能存活,造成致死性突变(lethal mutagenesis)或者错误灾难(error catastrophe)。添加NI后,病毒每复制一次,适合度都降低,最后,NI会耗尽核苷酸库[20]。
正链RNA病毒基因组复制的代时短,同源和非同源重组多,错误率高,病毒产量高,子代是一堆适合度各异的突变病毒。适合度高的耐药病毒株容易出现,抗病毒药物容易失效。因为Nsp14-exoN依赖RNA的RNA 3'-5'外切核酸酶校读活性,冠状病毒的复制忠实性比其他RNA病毒高20倍。
利巴韦林(也称病毒唑,结构式1-β-DRibofuranosyl-1,2,4-triazole-3-carboxamide)是鸟苷类似物,抑制冠状病毒mRNA合成,具有广谱抗病毒活性。单磷酸形式的利巴韦林与宿主合成核苷酸的关键酶-次黄苷酸脱氢酶(inosine monophosphate dehydrogenase,IMPDH)相互作用,减少核苷酸产生,耗尽细胞GTP库,抑制病毒RNA合成。病毒聚合酶掺入利巴韦林的三磷酸形式则导致致死性突变。体外,高剂量利巴韦林可以部分抑制MERSCoV和SARS-CoV复制。体内,利巴韦林则提高了病毒载量,加剧了SARS-CoV感染小鼠的病情。但是,对于SARS-CoV患者,利巴韦林没有效果,反而毒性很大。冠状病毒天然耐受5-氟尿苷和利巴韦林与nsp14有关。ExoN的校读活性可以使对其他病毒有效的利巴韦林失效。如何使得NI逃避Nsp14-exoN识别或者RNA延伸速度超过Nsp14-exoN外切速度,都是克服Nsp14-exoN挑战的有利方法。
低剂量利巴韦林和IFN-α2b具有协同效果,体外和在恒河猴中的效果都较好,但是对危重MERSCoV感染者的临床结局无改善。MERS-CoV感染14d使用利巴韦林和IFN-α2a可以提高生存率,但28d时施用则没有效果。但是也有研究发现利巴韦林和IFN-α2a或者IFN-β1联用未提高MERS患者生存率。IFN-α2b和IFN-β1b也用于MERS-CoV确诊患者治疗。IFN-β1a和利巴韦林联用对确诊者生存率无改善。并发症和治疗起始时间,可能影响个体疗效。有利的治疗时间窗口一般是感染后48h。HIV天冬氨酸蛋白酶抑制剂lopinavir/ritonavir和利巴韦林可以降低医疗人员MERS-CoV感染率约40%。lopinavir/ritonavir和IFN-β1b联合治疗,可以改善感染MERSCoV的小长尾猴的症状和病理。2016年启动了lopinavir/ritonavir和IFN-β1b联合治疗MERS-CoV感染患者的2/3期 临床试验(NCT02845843),评估其安全性、有效性和可能性。但结果未知。
Beta-D-N4-hydroxycytidine(NHC)是胞嘧啶类似物。其机理主要是导致病毒突变,但是对冠状病毒抑制机理尚不清楚。腺苷酸类似物(Adenosine analogue)BCX4430,gemcitabine hydrochloride是脱氧胞嘧啶类似物,抑制MERS-CoV和SARS-CoV。尿苷类似物(Uridine analogue)6-azauridine抑制HCoVNL63。鸟苷类似物阿昔洛韦抑制HCoV-NL63和MERS-CoV。
免疫抑制剂咪唑核苷类抗代谢药咪唑立宾(Mizoribine)可抑制嘌呤合成途径中的次黄苷酸脱氢酶(IMPDH)和单磷酸鸟嘌呤核苷合成酶(cMP),使鸟苷酸合成减少,细胞内RNA和DNA合成减少,抑制SARS-CoV,但是也阻止增殖的淋巴细胞由G0期进展为S期,抑制抗体的产生及记忆性B淋巴细胞和记忆辅助性T淋巴细胞的产生。
抗体:单克隆抗体(monoclonal antibodies,mAbs)领域正在飞速前进。单克隆抗体亚型包括IgG1,IgG1κ mAb等。目前主要开发靶向病毒保守结构或者膜蛋白的广谱中和单克隆抗体。但是,单克隆抗体的逃逸突变体也很常见。单克隆抗体需要考虑的方面包括:抗体亚型、半衰期、活性、组织滞留时间、不同部位的药代动力学、剂量、给药途径(口服、注射等)、与其他药物的联合使用、患者耐受性、抗原性等。单价、三价或多价纳米抗体也需要考虑。
转基因牛生产人多克隆IgG抗体:靶标是MERSCoV刺突蛋白(spike protein)(SAB-301)。体外,该多克隆抗体可以有效中和病毒,降低被感染小鼠肺部的病毒滴度。I期临床(NCT02788188)结果发现SAB-301安全性和耐受性较好。针对这个靶标的单克隆抗体,也在进行单药剂量增加的静脉注射的安全性、耐受性、药代动力学和免疫原性评估(NCT03301090)。
3.2 其他药物
IFN-αβ抑制剂和IFN-λIFN-αβ诱导ISGs,限制病毒复制。IFN-αβ招募更多IMM和其他免疫细胞发挥功能,也会加剧病情。SARS-CoV-感染小鼠中,早期干扰素应答有保护作用,滞后IFN-αβ信号传递使得抗SARS-CoV免疫应答失控。IFN治疗的时机很重要。治疗时机正确才可能有效。IFN-αβ受体阻断剂或者拮抗剂可防止晚期重症患者的过度炎症。IFNαβ主要通过单核细胞-巨噬细胞介导促炎症活性。与IFN-αβ不同,IFN-λ主要激活上皮细胞的抗病毒基因,又不过分刺激免疫系统,阻止招募嗜中性粒细胞到炎症部位。 SARS-CoV和MERS-CoV主要感染肺泡上皮细胞,因此IFN-λ可能用作治疗。
抑制氧化磷脂(oxidized phospholipid,OxPL)[21]:甲型流感病毒(influenza A virus,IAV)感染的小鼠中,氧化磷脂经TLR4-TRIF信号通路,增加肺部巨噬细胞的细胞因子/趋化因子产生,促进急性肺损伤(acute lung injury,ALI)。TLR4拮抗剂Eritoran(分子式C66H126N2O19P2)降低OxPL、炎症性细胞因子和趋化因子水平,保护小鼠免受IAV致死性感染。Eritoran免疫调控活性强,但无直接抗病毒活性,可以降低炎症应答。Eritoran或其他类似化合物抑制OxPL,抑制hCoV诱导的炎症。

表3 冠状病毒感染防控措施
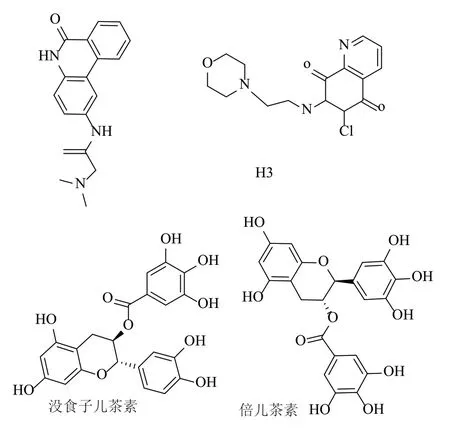
图5 部分化合物结构
鞘氨醇-1-磷酸受体1(Sphingosine-1-phosphate receptor 1,S1P1)拮抗剂:感染IAV的小鼠,内皮细胞鞘氨醇-1-磷酸受体1信号传递可以协调病理性炎症应答。靶向S1P1激活可以限制过度招募炎症细胞,抑制促炎症细胞因子和趋化因子,降低IAV诱导的发病率和死亡率。SARS-CoV感染人和非人灵长类的肺部上皮细胞和内皮细胞,驱动S1P1-介导的炎症性细胞因子/趋化因子应答,积累嗜中性粒细胞和巨噬细胞。激活S1P1可能治疗hCoV患者,降低病理性细胞因子/趋化因子应答。
3.2.1 抑制单核细胞招募和功能的分子
IMMs在致死性hCoV感染中发挥功能。小鼠心肌炎症模型中,脂类纳米颗粒递送含有干扰CCR2的RNA(short interfering RNA,siRNA)可以降低CCR2 mRNA,破坏招募IMM到炎症部位的功能,改善预后。hCoVs是单链RNA(single-stranded RNA,ssRNA)病毒,TLR7激活剂R837 (合成的ssRNA模拟分子)刺激IMMs,诱导很强的炎症应答。这提示hCoV感染时,可能是IMM特异性TLR-7信号传递促进过度炎症。TLR7拮抗剂可能减轻炎症,改善治疗。
3.2.2 其他免疫调节分子
hCoV感染后,可能减轻炎症应答的免疫调节分子包括细胞因子/趋化因子抑制剂,危险相关分子模式(danger-associated molecular pattern,DAMP)拮抗剂。SARS-CoV感染小鼠中,TNFα是急性肺损伤的重要因素,破坏T细胞应答。体外中和TNFα活性或者感染缺乏TNFR的小鼠,都可以预防SARS-CoV-诱导的发病和死亡。但是,TNFα只在SARS感染者后期血清中检测到。
炎症是有效免疫应答必不可少的,否则很难清除致病菌。首先是识别致病菌,然后招募免疫细胞,清除致病菌,最后修复组织,恢复稳态。但是,高致病性冠状病毒等诱导大量持久的细胞因子/趋化因子,称为细胞因子风暴,导致免疫病变和高死亡率。尸检提示来自IMM和嗜中性粒细胞的细胞因子/趋化因子具有重要作用。干预细胞因子/趋化因子可能减轻病理性炎症应答。感染早期、晚期的高病毒载量与人疾病严重程度相关,控制病毒载量、降低其炎症应答可能也有利于治疗。这需要找出冠状病毒感染患者或者动物中,介导炎症应答的特异性信号途径。
4 有待研究的问题
病毒颗粒相关的附属蛋白在冠状病毒生活周期中的作用?附属蛋白抑制干扰素信号传递的机制?附属蛋白与病毒结构蛋白之间的相互作用?不同冠状病毒入侵方式及主要功能受体差异?不同冠状病毒S蛋白的裂解特点及诱导膜融合规律?病毒组织嗜性与致病性改变同S蛋白结构的关系?CoV nsp15 EndoU的天然靶标?EndoU活性对dsRNA命运的影响?如何调控EndoU活性,避免误切?EndoU如何逃避宿主dsRNA感受器的免疫识别?与nsp15相互作用的宿主细胞/病毒的分子如何调控EndoU活性?冠状病毒等RNA病毒基因组复制的场所是所有地方还是局限在双层膜复制体?与其复制有关的各种膜结构的蛋白质成分与构象,是否有差异?形成复制细胞器时,病毒和宿主的蛋白质如何相互作用?双层膜(DMV)中积累的RNA,有何变化?宿主控制免疫应答网络的SNP及其在控制冠状病毒感染中的作用。从表观遗传角度如甲基化、泛素化、磷酸化、糖基化、乙酰化、染色质重塑和非编码RNA等研究人体应对CoV感染的调控,以及CoV逃避宿主的免疫反应的表观遗传调控,从SARS-CoV等冠状病毒感染细胞的转录组可否揭示病毒复制的信号通路和网络?动物模型与人体之间的差异,探索病毒侵入、感染机制、免疫机制、需要病毒免疫学、预防医学、药学和临床治疗学等领域协作。
