X-连锁迟发性脊椎骨骺发育不良一家系TRAPPC2 基因变异分析
2020-06-06王莉莉吴海瑛王晓艳张丹丹谢蓉蓉王凤云陈秀丽陈临琪
王莉莉 吴海瑛 孙 辉 王晓艳 张丹丹 谢蓉蓉 王凤云 陈 婷 陈秀丽 陈临琪
苏州大学附属儿童医院内分泌遗传代谢科(江苏苏州 215000)
X-连锁迟发性脊椎骨骺发育不良(X-linked spondyloepiphyseal dysplasia tarda,SEDT-XL,OMIM313400)是一种X-连锁隐性遗传性骨骼疾病,其特征是椎体和/或长骨骨骺结构缺陷,导致不成比例的身材矮小和关节过早退化。SEDT-XL通常在男性发病,女性为致病基因携带者,表型无异常。受影响的男性出生时的身长和身体比例无异常,在6~8岁左右开始出现生长迟缓,成年平均身高<150 cm[1]。国外报道的人群发病率为1.7/100万[2]。本研究采用全外显子测序及Sanger测序相结合的方法,分析1个家系的TRAPPC2基因,发现TRAPPC2基因4号外显子区域c.115delC缺失变异,现将结果报告如下。
1 临床资料
先证者,男,9岁2个月,因生长缓慢就诊,语言运动发育无落后,智力发育无异常。身高115 cm(<-3SD),臂间距109 cm,上部量56 cm,下部量59 cm,体质量21 kg,招风耳,尖下颌,牙列不齐,颈短,脊柱侧弯,心肺腹未见明显异常。患儿系G1P1,足月顺产,出生体质量3.5 kg,无窒息抢救史,无慢性疾病史。骨龄相当于8~9岁。脊柱X线正侧位片示脊柱侧弯,椎体扁平,椎体前部上下缘凹陷、中后部呈驼峰样突起改变(图1)。父母非近亲结婚,体健,父亲165 cm,母亲 3155 cm;舅舅身高132 cm,臂间距153 cm,无其他相关临床表现。
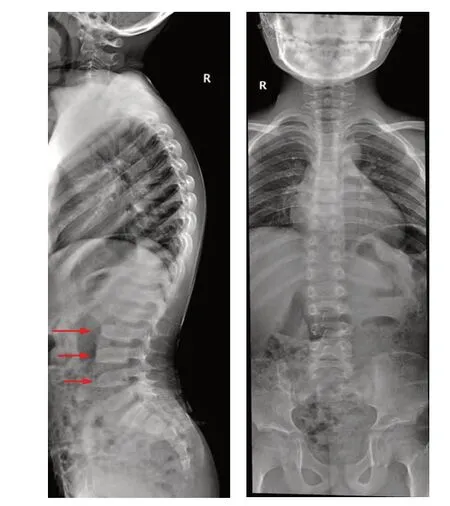
图1 先证者脊柱正侧位片
为明确病因,经医学伦理审核及家长知情同意后,抽取先证者及其父母和舅舅外周血各2 mL,提取DNA,构建DNA 文库,进行DNA 样本捕获;取捕获后的DNA 样本进行Illumina novaseq高通量测序,并通过Sanger 测序进行验证。测序数据经Illumina Sequence Control Software评估合格后进行数据读取和生物信息学分析。结果显示,在先证者TRAPPC2基因4 号外显子区域发现1 个缺失变异,c.115 delC,导致氨基酸改变p.Q39Sfs*3。通过Sanger测序验证与高通量测序结果一致,为半合子突变。先证者母亲为该突变携带者,父亲未检测到该突变,先证者舅舅存在相同的半合子突变位点(图2)。根据2015年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异分类标准,该变异为致病性变异(PVS 1+PM 2+PP 1+PP 4)。经检索PubMed、HGMDpro数据库、万方数据库、中国知网等国内外文献数据库,突变位点c.115delC未见报道。
2 讨论
TRAPPC2是SEDT-XL唯一涉及的基因,定位于Xp22,编码一种由140个氨基酸组成的蛋白质,包含6个外显子,跨越约20 kb的基因组区域[3]。文献报道TRAPPC2基因突变发生在由外显子3~6组成的整个编码区域,其中约90%涉及外显子4、5和6[4]。目前该基因功能尚不完全清楚。有研究表明,TRAPPC2蛋白定位于细胞核周围,可能参与软骨细胞内质网到高尔基复合体之间的物质运输,因此对于维持软骨细胞外基质的稳态至关重要[5]。
SEDT-XL的主要临床特点为不成比例的身材矮小,颈短,常伴有桶状胸、脊柱侧弯;在成年期会出现继发性骨关节炎,表现为腰背部疼痛、大关节活动受限;严重患者在40岁左右需进行关节置换术。典型的SEDTXL影像学表现为椎体前部上下缘凹陷,中部呈驼峰状,椎间隙狭窄,股骨头扁平,股骨颈相对较短[6]。本例先证者的主要表现为身材矮小,颈短,脊柱侧弯;影像学改变与文献报道一致。先证者舅舅,26 岁,身材矮小(132 cm),现尚未出现腰背部疼痛、关节活动受限等异常表现。根据先证者的临床表型、典型的影像学特征、家族史,及基因检测结果考虑TRAPPC2基因外显子4 的c.115 delC 突变为致病变异。既往该变异位点未见报道。曾有报道2例SEDT-XL,1例为起始密码子的错义突变,另1 例为c.40 delG 缺失突变,均导致蛋白翻译提前终止,并通过体外实验证明TRAPPC 2 失功能是导致SEDT-XL 表型的主要原因[7]。其详细的致病机制尚不明确,有待进一步功能验证。
2015年国内报道首例TRAPPC2基因第3内含子的剪接供体c.93+5 G>A 突变,并总结了中国人群中已发现的12种TRAPPC2基因突变类型,缺失和剪接突变各4 个、错义突变2 个、无义突变和插入突变各1个[8]。2015年8月国内发现首例TRAPPC2基因第5外显子c.271-275delCAAGA缺失突变[9]。本例先证者TRAPPC 2为新的缺失变异,丰富了中国人群SEDTXL的基因突变谱。到目前为止,在人类基因突变数据库中已报道的TRAPPC2基因变异类型达57种,包括32 个插入或缺失突变、10 个剪接突变、9 个无义突变和6 个错义突变。大多数突变会导致蛋白翻译终止,TRAPPC2功能缺失[7]。
TRAPPC2突变的基因型和表型之间的相关性尚未明确,但有研究者认为,随着TRAPPC2基因突变位点越靠近5 '端,临床表型越重,而越靠近3 '端,其临床表现有减轻的趋势,位于TRAPPC 2基因第5~6 外显子突变的临床表型往往较轻,患者常无或有轻微的髋关节疼痛,而位于第3~4外显子的突变则会引起脊柱畸形,并相对较早出现关节疼痛及较为严重的并发症[1.10]。本家系的TRAPPC2基因变异位于第4外显子区域,编码第39个氨基酸的位置,靠近5 '端,先证者及其舅舅均表现为身材矮小,尚未出现关节疼痛表现,与文献报道存在一定偏差,考虑可能与先证者及其舅舅年纪尚轻有关,仍需进一步随访,具体的机制有待进一步研究。有研究者认为,TRAPPC2基因变异临床表型的异质性可能与其伪基因相关[7]。已发现的伪基因有9个,最有可能的是TRAPPC2B(也称为SEDLP1),位于19q13.4上,具有转录活性,编码与TRAPPC2基因编码相同的蛋白质,可能与SEDT-XL患者临床表型异质性相关。文献报道称,TRAPPC2基因变异患者若存在伪基因表达,临床表型可能相对较轻[7.11]。
SEDT-XL目前尚无有效的治疗方法。对于SEDTXL引起的矮小,使用生长激素治疗的研究尚不足。国内报道1例以矮小就诊的SEDT-XL患儿,予生长激素治疗1 年,效果不佳[12]。患儿应尽量避免可能损伤脊柱及大关节的活动,以延缓疾病的进展,SEDT-XL多在中年期出现严重的腰背部及关节疼痛、活动受限,需进行关节置换术[13]。
基因检测是SEDT-XL确诊的重要工具,还有助于检测出携带者,为患者家庭提供遗传咨询及产前诊断,减少该疾病患儿的出生。由于SEDT-XL一般在儿童中晚期才开始出现典型症状,因此,对于儿童矮小合并脊柱畸形,且家系中有男性身材矮小者,应考虑是否患有此种疾病,必要时行基因检测明确诊断。早期干预可延缓疾病的进展。
