NBAS 基因缺陷症一家系报告及文献复习
2020-06-06吴非霏胡宇慧陈哲晖廖建湘陈淑丽
吴非霏 崔 冬 胡宇慧 陈哲晖 陈 黎 廖建湘 陈淑丽
中国医科大学深圳市儿童医院(广东深圳 518038)
NBAS基因缺陷症为一种罕见病,由NBAS基因变异所致,于2010年首次报道[1]。NBAS基因缺陷症的临床表现主要为早老面容、视神经萎缩、反复肝功能损害、身材矮小、免疫功能缺陷导致的反复感染等[1-3]。NBAS基因缺陷症临床诊断困难,确诊需依靠基因检测。本文回顾分析深圳市儿童医院收治的一家系2例由基因诊断明确的NBAS基因缺陷症患儿的临床资料,并复习相关文献进行分析。
1 临床资料
先证者,男,14岁半。身高124 cm(<-3 SD),体质量20 kg(<-3SD),轻度精神发育迟滞(韦氏儿童智力测试智商54分)。手脚小,皮肤松弛,皮下脂肪少;早老面容,三角脸,双眼外凸,小下颌,牙齿发育不齐;心肺未见明显异常;腹平软,肝脏肋下2 cm,质地中等,脾脏肋下未触及;神经系统未见明显异常。患儿为G1P1,38+2周牵引助产出生,出生体质量2 200 g,无窒息抢救史。母孕期健康状况良好,父母非近亲结婚。患儿于1月龄时诊断斜颈、髋关节发育不良;6月龄时发现生长发育落后于同龄儿,1 岁后开始多病,多为上呼吸道感染,2岁时只会说“爸爸、妈妈”、开始独立行走。先证者自2岁起监测天门冬氨酸氨基转移酶波动于61~1 261 IU/L,丙氨酸氨基转移酶波动于120~1619 IU/L,血乳酸波动于2.47~3.2 mmol/L,血氨波动于16~93 μmol/L;免疫球蛋白IgG 3.19 g/L,IgM 0.35 g/L,IgA<0.244 g/L;铜蓝蛋白42.2 mg/dL(正常值15~30 mg/dL);血涂片可见Pelger-Huët细胞(图1)。心电图示窦性心动过速并窦性心律不齐。左腕X线正位片示骨龄在正常范围;骨盆X线正位片示先天性髋关节脱位。患儿2月龄超声示右侧胸锁乳突肌结节、右侧骨性髋臼发育不良、左侧髋关节半脱位。2.5 月龄时头颅CT 示蛛网膜下腔增宽、脑白质偏少。14 岁半眼科检查示视神经萎缩,视力左眼4.6、右眼4.6,红绿色盲。心脏超声、颅脑MRI均未见明显异常。

图1 患儿血涂片典型Pelger-Huët 细胞(箭头所示)
先证者妹妹,7岁11个月,身高100 cm(<-3SD),体质量11 kg(<-3 SD),学习成绩较同龄儿童落后,容貌外形与先证者相似。G2P2,36周顺产,出生体质量2 200 g,无窒息抢救史。母孕期妊娠期糖尿病史。患儿于16 月龄时可独立行走。出生后监测肝酶,天门冬氨酸氨基转移酶波动于34~11 280 IU/L,丙氨酸氨基转移酶波动于48.5~4 185 IU/L,血乳酸波动于2.4~3.35 mmol/L,血氨波动于40~66.4 μmol/L;IgG 3.49~3.55 g/L,IgM 0.17~0.3 g/L,IgA < 0.0667 g/L;凝血酶原时间 36 s(正常值9.3~12.9 s),凝血酶原时间(INR)2.49 INR(正常值0.72~1.15 INR),余未见异常。血涂片可见Pelger-Huët细胞。染色体核型分析46,XX,1qh+(属遗传多态性)。1岁时颅脑CT未见明显异常。6岁时肝脏超声示肝脏右肋下2.5 cm。7岁11个月眼科检查示视神经萎缩,双眼视力左眼4.5、右眼4.5,红绿色盲。
经医学伦理审核,家长知情同意后行基因检查。先证者及其妹妹的外周血样被送至北京金准基因科技有限责任公司进行全外显子测序以及一代测序验证。结果显示2 例患儿均携带NBAS基因复合杂合变异,变异位点为c.5752 A>C 和c.1599+1 G>C,其中位点c.5752A>C 来自其父亲,位点c.1599+1G>C 来自其母亲(图2)。患儿父母无相关临床表现。其中位点c.5752A>C为第5752号核苷酸上腺嘌呤替换为胞嘧啶,c.1599+1 G>C为第1599 号核苷酸下游一位的内含子核苷酸由鸟嘌呤替换为胞嘧啶。c.5752 A>C(p.T 1 9 1 8 P)在正常人群中未见携带(P M 2),MutationTaster[4]、SIFT,PROVEAN[5]、Polyphen2[6]均预测为有害(PP3);该位点首次被报道于1例携NBAS基因复合杂合变异的SOPH 综合征患者中(PM 3),并报道为致病变异位点(PP 5)[7]。根据ACMG 指南,该变异被评为疑似致病性突变(likely pathogenic,PM 2+PM 3+PP 3+PP 5)。而c.1599+1 G>C 为新发现的变异位点,该位点变异使NBAS基因第15 号外显子和第15 号内含子交界处的碱基发生变化,根据MutationTaster[4]和Human Splicing Finder[8]的预测,该变异直接影响15号内含子的剪切,为恶性剪切突变(PVS 1)。该变异在正常人群中未见携带(PM 2),且其反式等位基因上存在已被报道的致病变异(PM3)。根据ACMG指南进行评级,这个剪切变异被评为致病性变异(pathogenic,PVS1+PM2+PM3)[9]。
先证者及其妹妹均确诊为NBAS基因缺陷症。2例患儿在感染时有肝酶增高,予抗感染、护肝等对症治疗,感染控制后,肝酶可自行下降,现可正常上学,成绩较同龄儿童落后。两例患儿现生长发育仍较正常同性别同龄儿落后,身高、体质量均<-3SD,轻度精神发育迟滞。
2 讨论
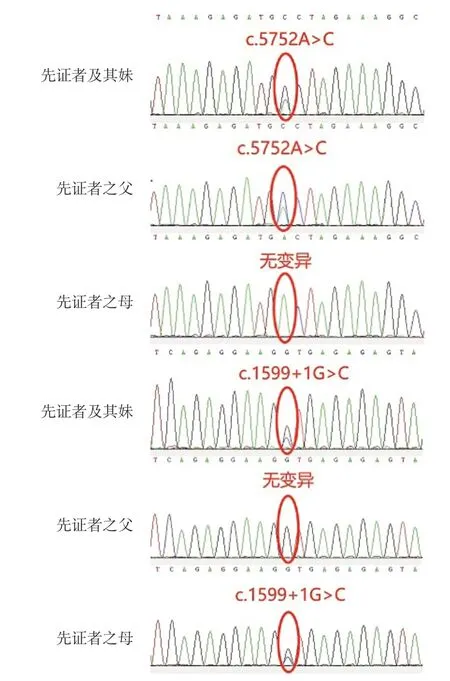
图3 患儿及父母NBAS 基因测序图
NBAS基因缺陷症为一种罕见病,人群中具体发病率不详[3]。NBAS基因位于2 p 24.4[10],表达长为2 371 个氨基酸的功能蛋白。该蛋白是介导膜运输的syntaxin 18复合体的一个组成部分,参与了高尔基体与内质网之间的逆转运过程,作用是平衡细胞内从内质网到高尔基体的分子运输[1,11-13]。同时,NBAS基因参与形成无义介导的mRNA 降解通路,即NMD(nonsense-mediated mRNA decay)通路。NMD通路的作用是识别和降解含提前终止密码子的转录本,也就是清除错误的功能蛋白,从而调节细胞应激反应和膜转运。NBAS功能的缺失可能会影响其在NMD通路中的作用,影响其作为syntaxin 18复合体组成部分的活性[1,11-12]。
在NBAS蛋白中,第1~1 035号氨基酸为与囊泡转运蛋白USE 1 L 结合的功能域,第1 036~2 371 号氨基酸为与着丝粒/动粒同源蛋白ZW10(Zeste white 10)以及调控蛋白RINT1(RAD50-interacting protein 1)结合的功能域[13-14],这两个功能区间均对NBAS蛋白介导胞内膜系运输并通过NMD通路清除错误蛋白起重要作用。在本组患儿的复合杂合变异中,c.5752A>C变异使1 918 号的苏氨酸突变为脯氨酸,影响蛋白与ZW 10 和RINT 1 结合的功能;c.1599+1 G>C 新变异影响第15号内含子的剪切,打乱533号氨基酸以后的全部序列,使突变蛋白完全失去功能。该复合杂合变异导致患者体内缺乏功能性NBAS蛋白,影响胞内膜系的正常运输以及NMD通路的正常调控。
NBAS基因缺陷症引起肝脏病变的机制可能与如下三方面有关:①当NBAS基因缺陷时,syntaxin 18复合体的热敏感性增加,导致内质网和高尔基体间的转运功能受到干扰[7,11]。②NBAS基因下调导致CCL20和IL6R过表达,其中CCL20是脂多糖诱导的肝脏炎症的生物标志物及中介物,CCL20过表达可能促进应激因子引起肝脏炎症和细胞溶解[1,15]。③NBAS基因下调导致IL6R过表达,IL6参与机体的免疫调节,NBAS基因缺陷可能导致IL6R mRNA降解降低,使得肝细胞对炎症刺激因子的敏感性增加,这可解释肝脏病变与免疫学的联系[1]。本研究2例患儿在感染引起发热时均有明显的急性肝损害,肝酶明显升高,而当感染控制、体温正常时,肝酶随之明显下降。但尽管急性感染发热时,患儿的肝酶可升高至上千甚至上万,而患儿精神状态尚可,无黄疸和凝血功能障碍。肝酶上升的幅度与临床表现并不相符,原因有待进一步研究。
NBAS基因下调可能导致γ-羧基谷氨酸蛋白(MGP)过表达,MGP调控骨骼形成。MGP的过表达将导致身材矮小和骨骼缺陷[1,11,16-17]。本文先证者存在身材矮小和先天性髋关节脱位,其妹妹仅有身材矮小。
NBAS基因缺陷症引起的视神经萎缩通常由能量代谢紊乱导致,视网膜营养不良可能与高尔基-纤毛囊泡转运紊乱有关,故认为NBAS基因可能在视网膜稳态中发挥重要作用[3]。本文2例患儿均有视神经萎缩。
NBAS基因缺陷可累积多个系统[18],临床上主要导致两种情况,一种是婴儿期反复发作的急性肝衰竭,另一种是SOPH 综合征[1,16,19]。由NBAS基因变异引起的婴儿期反复发作的急性肝衰竭通常无肝脏基础疾病[20],常在急性感染性疾病的诱发下,出现轻中度黄疸、丙氨酸氨基转移酶及天门冬氨酸氨基转移酶显著增高、凝血功能异常等;亦有部分患儿可有低血糖、高氨血症、高乳酸血症,甚至可发展为肝性脑病;少数患者可并发心肌病、自身免疫性胃肠疾病和癫痫等[21-22]。由NBAS基因变异导致的SOPH综合征(身材矮小、视神经萎缩、Pelger-Huët畸形综合征)为常染色体隐性遗传[2],主要临床表现包括生长发育迟缓,早老面容、皮肤松弛、双眼外凸、视神经萎缩导致视力丧失及色盲、骨质疏松等[1,3,23]。血液及骨髓涂片中可见典型的Pelger-Huët 细胞,肝酶可正常或增高,可有低丙种球蛋白血症,故易患各种感染[2,10,18,24]。本文2 例患儿既有明显肝损害的表现,又有SOPH 综合征的临床表现。
NBAS基因缺陷症的明确诊断主要依靠基因诊断。当患儿出现早老面容、视神经萎缩、婴儿期反复发作的肝功能损害、生长发育迟缓等多系统异常,实验室检查出现反复的肝酶升高,血液及骨髓涂片中发现典型的Pelger-Huët细胞时[1-2],需考虑本病并行相关基因检测。
NBAS基因缺陷症无特殊的治疗方法,主要为对症治疗。当患儿出现发热、感染等引起的急性肝损害时,应积极予抗感染、护肝及营养支持治疗。有研究表明,应用辅酶Q和左卡尼汀可减少急性肝损害发作频率及持续时间[11-12,25]。当患者出现反复发作的肝衰竭时,亦可考虑行肝移植[7]。当患者同时合并低丙种球蛋白血症时,予丙种球蛋白静脉输注可有效减少感染发生[2]。也有研究称应用生长激素可改善患儿身材矮小,进而改善由矮小引起的心理健康问题[26]。
NBAS基因缺陷症虽没有特殊治疗方法,但随着年龄增长,患儿的肝功能会有明显改善,肝衰竭发作频率明显减少,但身材矮小无明显改善。患儿智力发育水平处于边缘状态,生活质量尚可,但需要密切关注患儿因早老面容和明显矮小等所引起的心理健康问题。
