KBG 综合征1 例报告及文献回顾
2020-06-06曹玉红张立毅曹开方张光运
曹玉红 张立毅 曹开方 张光运
1.空军特色医学中心(北京 100142);2.北京大学医学部(北京 100191);3.空军军医大学口腔医院 (陕西西安 710032)
KBG综合征(KBG syndrome,KBGS)是一种罕见的常染色体显性遗传病,主要表现为独特的颅面特征、巨齿畸形、身材矮小、智力残疾和骨骼异常等[1-6]。KBGS 由锚蛋白重复结构域蛋白11(ankyrin repeat domain-containing protein 11,ANKRD11)基因突变或包含ANKRD11在内16q24.3的缺失引起[1,3-4]。到目前为止,国外已报告约200例,国内报告1例[3]。现回顾性分析1例经基因诊断确诊的KBGS患儿的临床资料,并检索相关文献,探索KBGS 的临床及基因突变特点。
1 临床资料
患儿,男,1 岁11 个月,以智力运动发育落后、耳聋及间断抽搐1年余入院。患儿为G1P1,足月顺产,无窒息史。生后8 个月独坐,1 岁6 个月独走。不能言语,听力差,耳鼻喉科诊断为耳聋。近1年抽搐3次,表现为意识丧失,双眼凝视,四肢阵挛抽搐,持续1~5分钟,诊断癫痫,服用丙戊酸钠治疗。1岁半时行睾丸鞘膜积液手术。家族中无类似疾病患者。体格检查:体质量9.8 kg,身高79 cm(<P3),头围45.5 cm。特殊面容:眼睑下垂,眼距宽,内眦赘皮,耳廓及鼻梁突出,球状鼻,长人中,薄上唇,中切牙宽大畸形。胸骨左缘第二肋间闻及Ⅲ级连续性杂音。四肢肌张力低。脊柱无畸形,双手手指短,小指向桡侧弯曲,双手通贯掌纹(图1)。实验室检查:甲状腺功能、有机酸及酰基肉碱谱分析正常。染色体核型:46,XY,染色体未见异常。X线片示左腕关节骨化中心未出现(图1)。超声心动图示动脉导管未闭。脑电图示双侧顶、枕导痫性放电。头部核磁共振成像未见异常。“0~6岁智能发育筛查量表”测试结果:发育商(DQ)<70,相当于16.5个月,智力指数(MI)<70,相当于18个月。患儿父母表现无异常。
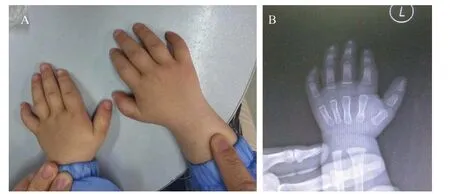
图1 患儿手外观及X 线片表现
患儿临床诊断疑似KBGS,为进一步明确诊断,经医学伦理审核以及家长知情同意,对患儿及其父母进行外显子基因组检测。采用QIAamp Blood DNA Mini Kit 试剂盒(德国Qiagen 公司)提取有核细胞基因组DNA。采用SureSelect Human All Exon Kit Vl试剂盒(美国Agilent公司)捕获建库,最后使用Illumina HiSeq 2500 测序系统进行高通量测序。测序数据经Illumina Sequence Control Software 评估合格后,应用NextGENe 软件(美国SoftGenetics 公司)读取数据,Ingenuity Variant Analysis 软件(美国Ingenuity Systems 公司)进行生物信息学分析。结果发现:患儿ANKRD 11基因基因6 号外显子存在一处杂合突变c.316 C>T (胞嘧啶>胸腺嘧啶),致氨基酸变化p.R106X(精氨酸>终止密码子)。该变异的染色体位置为chr 16:89357502。最后用Sanger 测序方法验证ANKRD11基因变异。扩增ANKRD11基因6号外显子的引物序列:正向引物 5'-GTA AGG ACC TAG CTT TGG AG -3',反向引物为5'-TGT CCA ATC TTC AAG AGC CC -3'。PCR 产物纯化后进行Sanger 测序及数据分析。结果示患儿ANKRD11基因存在杂合突变,父母双方均未检出该突变,患儿突变为新发突变(图2)。此突变在 HGMD 及Clinvar 数据库中无收录,该位点的人群频率无收录。在gnomAD 数据库的东亚人群中无收录。MutationTaster 预测为 A(评分1.0)。ACMG相关基因突变解读指南分级评定为Pathogenic。患儿最终诊断为KBGS。
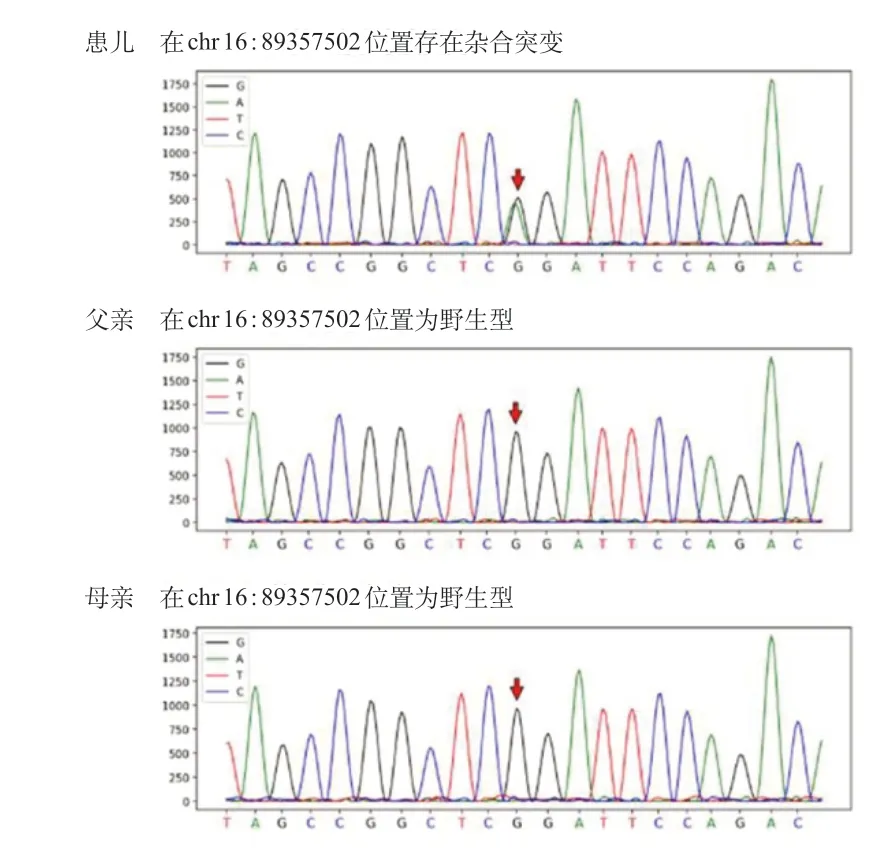
图2 患儿及父母ANKRD11 基因外显子基因组测序图
2 讨论
KBGS 是一种多发性先天性异常综合征,病变可累及全身多器官系统[1-6]。KBGS 于1975 年首次被报道,其疾病名称来源于最早诊断的三个家庭姓氏的首字母[7]。2011 年确定位于16 号染色体长臂的ANKRD11基因为KBGS的致病基因[8]。以“KBG综合征”、“ANKRD11”为关键词检索维普数据库、万方数据库及中国知网数据库自建库至2019年12月收录的文献,检索到ANKRD 11致KBG 综合征的相关文献1篇[3]。以“ANKRD11”、“KBG syndrome”、“16q24.3 microdeletion”为关键词检索PubMed、Web of Science数据库自建库至2019 年12 月收录的文献,共检索到28篇文献[3,5,9-13],报道KBGS综合征患者200例,发病年龄1~66岁,男女比例1.5∶1。KBGS患者ANKRD11基因内单核苷酸突变和小的插入缺失突变约占致病性突变的71%,包含ANKRD11的16q24.3缺失约占29%,没有突变热点[5]。ANKRD11基因内突变患者中66%为新发突变,16q24.3缺失患者中75%为新发突变,其余患者为常染色体显性遗传[4]。研究证实,较大范围16q24.3缺失(包括ANKRD11和周围基因)的患者有更严重的临床表现,除此之外,基因型与表型之间无明确的相关性[4]。女性患者症状相对较轻。据统计,99%的KBGS 患者有颅面部畸形,面部呈三角形、短头畸形、一字眉、宽眉毛、眼距宽、内眦赘皮、上睑下垂、耳廓及鼻梁突出、球状鼻、鼻孔前倾、长人中、薄上唇等[5]。约84%的患者有巨齿畸形,尤其是上中切牙,此外尚可见到裂齿、尖牙、牙釉质发育不全、牙齿拥挤、少牙和多生牙等。少数患者可有腭裂、悬雍垂裂和腭咽发育不全[5,14]。90%的患者有轻到中度智力迟缓,动作语言发育落后[5]。约半数患者脑电图异常,伴或不伴癫痫发作,抗癫痫药物治疗大多有效,部分患者在青春期后癫痫发作有所缓解[11]。少数成年患者可生活自理或参加工作。大部分患者有神经影像学异常:如小脑、胼胝体、视神经等发育不良,Chiari畸形,Dandy-Walker畸形,松果体及蛛网膜囊肿,空洞脑,脑脊髓膜膨出,脑室周围白质软化及灰质异位等。60%的KBGS患者合并行为问题:如注意力缺陷多动障碍,孤独症谱系障碍,强迫,焦虑等[4-5]。约50%患者生后逐渐出现身材矮小,骨龄延迟。75%的患者合并骨骼异常,如颈肋、脊柱裂、脊柱后凸或侧凸、大前囟或前囟闭合延迟、髋关节发育不良、短肢、指弯曲以及并趾等[4-5]。32%的患者出现听力异常、复发性或慢性中耳炎。听力损失可以是传导性、感音神经性或混合性的[15]。KBGS患者常合并斜视、近视、先天性白内障、巨大角膜等眼部病变[4]。部分KBGS患者可合并泌尿、生殖、消化及心脏病变,如隐睾、肾盂重复、膀胱输尿管反流、胃食管反流、斜疝、室间隔缺损、动脉导管未闭等[4,16]。少数患者可有色素沉着、鱼鳞病、毛发异常、指甲营养不良、血细胞减少、骨髓增生不良、性早熟等。罕见合并白血病、横纹肌样肿瘤等恶性疾病报告[4-5]。
本例患儿有精神运动发育迟缓、癫痫、身材矮小、骨龄延迟、小指弯曲、动脉导管未闭、听力障碍等,具有典型面部特征,如眼睑下垂、耳廓及鼻梁突出、球状鼻、长人中、薄上唇、中切牙宽大畸形等,符合KBGS的临床特征。本例患儿ANKRD11基因存在杂合突变c.316 C>T,致氨基酸变化p.R 106 X,该突变位点在国内外未见报道。由于该变异6 号外显子第316 号碱基C 变为T,引起第106 号氨基酸由精氨酸变为终止密码子,氨基酸合成提前终止,产生截短蛋白,导致ANKRD11蛋白质功能丧失。本例患儿进一步扩展了ANKRD11的基因突变谱。
目前ANKRD11基因突变导致KBGS的具体致病机制尚未完全清楚。研究证实,ANKRD11突变的Yoda小鼠神经元增殖及神经发生减少,神经元定位异常,表现出颅面异常、头盖骨宽、脊柱后凸畸形、骨密度降低、孤独症样行为[1,17]。ANKRD 11 在小鼠大脑皮质发育中通过BDNF/TrkB 信号通路调节锥体神经元迁移和树突分化[17-18]。ANKRD11是控制组蛋白乙酰化和神经发育过程中基因表达的关键染色质调节剂。ANKRD 11 蛋白通过招募组蛋白脱乙酰基酶与p 160辅活化子和核受体复合物的相互作用,参与转录激活[1]。ANKRD 11 可影响核受体配体依赖性转录激活和肿瘤蛋白TP 53 的抑瘤功能。ANKRD 11 是一个候选的肿瘤抑制基因,有学者推测其单倍不足可能导致KBGS患者的癌症风险增加[19]。
KBGS 诊断主要依据患儿临床表现,结合分子遗传学检查,证实有ANKRD 11基因突变或包含ANKRD 11 的16 q 24.3 缺失即可确定诊断[1,4]。国内外关于KBGS临床诊断标准尚未达成共识。2007年提出KBGS临床诊断标准[20],满足以下4个或以上临床表现需考虑KBGS:①上中切牙巨齿畸形;②面部特征异常;③手部异常;④广泛发育迟缓、癫痫、智力低下等中枢神经系统受累;⑤骨龄落后显著;⑥肋椎异常;⑦身材矮小;⑧一级亲属患KBGS。2016 年对上述诊断标准进行修订[1],主要指标包括:上颌中切牙过大、矮小、复发性中耳炎伴或不伴听力丧失、一级亲属患KBGS;次要指标包括:手指短或相关手部异常、癫痫、隐睾、喂养困难、腭弓异常、孤独症、前囟大或闭合延迟。发育迟缓、学习障碍或明显行为异常的患者,合并2个主要指标或合并1个主要指标及2个次要指标者即可临床诊断KBGS。基因测序可检出大部分ANKRD11致病突变。对于染色体缺失和重复,主要应用靶向基因的缺失和重复分析、染色体微阵列或比较基因组杂交等方法[4-5]。
目前KBGS尚无特效治疗方法,主要是对症和支持治疗。合并先天性心脏病、生殖泌尿系统畸形的患者需手术治疗。KBGS患者癫痫发作需用抗癫痫药物治疗[1-2]。喂养困难的患儿可根据病情给予鼻饲管喂养或造瘘管喂养。身材矮小的患儿给予生长激素治疗,可显著增加身高。性早熟的患者,可给予促性腺激素释放激素类似物等抑制青春期发育。KBGS患儿的运动及语言迟缓,需要进行运动及语言康复训练。定期监测听力、视力、生长发育、青春期发育和认知发育情况,避免应用耳毒性药物[1,4-5]。
综上,临床对于有特殊面容、巨齿畸形、身材矮小、智力残疾和骨骼异常等的患儿,应考虑到本病的可能,并尽早进行遗传咨询、基因检测,以明确诊断。
