Xp/Yq染色体间不平衡易位的临床遗传学特征分析
2020-06-05蔡婵慧李怡胡晶晶李星李显筝
蔡婵慧,李怡,胡晶晶,李星,李显筝
人类性染色体是从一对同源染色体进化而来,性染色体通过某些片段的丢失、插入或者重排使得两者差异性逐渐增大,同源性逐渐减小从而获得了各自独特的功能,但XY染色体上仍存在同源序列[1],在减数分裂时能相互配对形成联会复合体。目前认为XY染色体间的易位是由于这些同源序列间的异常交换引起的[2]。XY染色体间的不平衡易位根据重复缺失片段等不同而产生不同的类型,不同的易位类型遗传效应、表型和预后不同,产前诊断采取的干预手段也不同。现报告一家系遗传的XY染色体间不平衡易位导致的Xp部分缺失、Yq部分重复的胎儿病例,并进行相关遗传学分析及文献复习。
1 对象与方法
1.1 研究对象 孕妇32岁,身高150 cm,体质量50 kg,G2P1A0,孕20+周(自然妊娠),平素月经规则,B超提示宫内单活胎,未发现明显异常,胎儿双顶径45 mm,胎心率148 次/min,唐氏综合征筛查低风险,21三体风险度1∶522,游离雌三醇(uE3)中位数低(0.12),妊娠前有糖尿病,目前行胰岛素治疗,妊娠前和妊娠期间无其他服药史及有毒、有害物质接触史。夫妇非近亲结婚,表型及智力正常,双方均为地中海贫血携带者,孕妇本人地中海贫血基因型为--SEA/aa(已排除非缺失型α地中海贫血),丈夫地中海贫血基因型为βIVS-Ⅱ-654/βN(已排除α地中海贫血)。育有一男孩,5岁,足月剖宫产,外院染色体检查结果:46,Y,i(X)(q10),皮疹(诊断为先天性鱼鳞病),隐睾,智力发育迟缓,语言发育障碍,生长发育略低于同龄儿(体质量14 kg,身高97.70 cm),活动与注意力失调,喂养困难,睡眠紊乱(间歇性的白天嗜睡和夜间失眠),心肺腹未见异常,心率105次/min,律齐,呼吸平顺,双肺呼吸音清,未闻及干湿罗音,腹平软,肝不大,脑电图未见异常,骨代谢未见异常,血常规提示小细胞低色素性贫血,微量元素测定提示铁含量降低,血清铁蛋白正常,学校体检提示双眼屈光异常,眼科检查双眼眼位正,眼球运动无受限,双眼球结膜无明显充血,角膜透明,晶体透明,右眼视力不合作,眼压13 mmHg(1 mmHg=0.133 kPa),左眼视力不合作,眼压176 mmHg。孕妇本次妊娠因uE3中位数低及曾育染色体异常儿于孕20+周行羊水穿刺产前诊断,并签署知情同意书。
1.2 方法
1.2.1 染色体微阵列分析使用QIAGEN 公司生产的QIAamp DNA Blood Mini Kit提取试剂盒对羊水细胞进行基因组DNA的提取,使用美国Affymetrix公司的CytoScan 750k芯片,对提取的羊水DNA进行酶切、连接、PCR扩增,产物纯化及片段化、标记及杂交,芯片洗涤后扫描。检测结果使用Chromosome Analysis Suite(ChAS;version 3.1.0.15)软件进行分 析, 最 终 结 果 判 读 参 照OMIM、UCSC、ISCA、DGV、DECIPHER等数据库。
1.2.2 染色体G显带/C显带分析将在B超引导下穿刺获得的20 mL羊水分别接种到2个原位培养瓶和2个60 mm圆形培养皿中进行原位培养,培养7 d后换液。观察2个原位培养瓶中细胞克隆达到15个以上后开始收获。经吉姆萨染色制片,计数15~20个克隆,每个克隆分析1~2个核型;结果依据人类细胞基因组学国际命名体系(ISCN 2013)对染色体核型进行命名。同时加做C显带。羊水G显带分辨率为350~420条带。
2 结果
2.1 染色体微阵列分析结果 本研究结果为:arr[hg19] Xp22.33p22.31(168,551-8,428,908)x0,Yq11.221q11.23(16,099,875-28,799,654)x2。提示胎儿发现2处染色体异常:①X染色体Xp22.33-p22.31位置发生缺失(红色),片段大小约8.3 Mb;②Y染色体Yq11.221-q11.23位置发生重复(蓝色),片段大小约12.7 Mb,见图1。
2.2 染色体G显带/C显带分析结果 本研究结果提示X染色体短臂末端携带未知来源的额外片段,该片段呈浓染固缩状,结合C显带结果,确定该深染片段主要为异染色质。
经羊水染色体G显带/C显带检查结合微阵列分析,确定该衍生X染色体为Xp/Yq染色体间不平衡易位所致,核型为46,Y,der(X)t(X;Y)(p22.3;q11.22),衍生的X染色体由重复的Y染色体长臂和短臂末端缺失的X染色体重排形成。为明确该衍生X染色体的来源,本研究对夫妇双方外周血染色体进行检测,同时对外院诊断为46,Y,i(X)(q10)的胎儿哥哥进一步在本院行外周血染色体及微阵列检测。结果发现胎儿父亲染色体核型正常;母亲携带有相同的一条衍生X染色体,染色体核型为46,X,der (X)t (X;Y)(p22.3;q11.22);经本院外周血染色体及微阵列检测分析,外院诊断为46,Y,i(X)(q10)的哥哥实际上与胎儿结果一致。至此,可以确定胎儿以及哥哥的衍生X染色体为母系遗传,核型为46,Y,der(X)t(X;Y)(p22.3;q11.22)mat,见图2。
3 讨论
本研究中Xp/Yq不平衡易位导致Xp末端缺失及Yq部分重复,男性患者为Yq的部分双体并Xp部分纯合缺失,而女性患者为Yq的部分单体并Xp部分杂合缺失。由于该衍生X染色体没有Y着丝粒的存在,易位的Yq主要为没有编码基因的异染色质区域,不包含性别决定基因(SRY基因),Y染色体异染色质的长度变化与任何表型结果无关,所以该Xp/Yq易位患者的表型主要依赖于Xp的缺失而不是Yq的重复。莱昂假说认为,通常情况下女性体细胞内的两条X染色体之一随机失活,但如果存在结构畸变的X染色体,则X染色体失活出现偏倚,当女性发生X染色体与常染色体的平衡易位,正常的X染色体优先失活,以避免常染色体上基因失活带来的严重后果,当女性存在涉及到X染色体缺失、重复或等臂染色体等非平衡结构畸变时,通常是结构异常的X染色体优先失活,这可能是由于进化过程中异常X染色体优先失活能降低由该类异常导致的不良后果[3]。受X染色体失活偏倚影响,Xp/Yq易位女性患者含短臂缺失的X染色体通常优先失活,除了身材矮小,一般表型正常,智力正常,生育能力正常。由于女性患者生育力不受影响,将衍生的X染色体传递给下一代,使孩子拥有相同衍生X染色体的风险为50%。男性患者X染色体短臂末端缺失导致Xp连续基因综合征,连续基因综合征由于相邻基因共同缺失产生复杂临床表型,具体表型主要根据缺失片段上包含的特定基因以及数量而变化[4]。

图1 染色体微阵列分析图

图2 染色体核型/家系/模式图
本研究中,X染色体Xp22.33-p22.31(Xp22.31/pter)位置发生缺失,片段大小约8.3 Mb,包含了41个已知基因,其中包括矮小同源盒基因(SHOX)、芳香基硫酸酯酶E基因(ARSE)、神经连接蛋白4X基因(NLGN4X)、类固醇硫酸酯酶基因(STS)和卡尔曼综合征基因1(KAL1)5个已知与特定疾病相关的基因。矮小同源盒(SHOX)基因存在于人类性染色体的Xp22.32和Ypl1.3的拟常染色体区域内并且逃避X染色体的失活,属于单倍剂量敏感性基因。SHOX基因单倍剂量不足与特纳综合征、Leri-Weill综合征(LWD)有关,主要特点是身材矮小、四肢中部发育不良如前臂和小腿短、脊柱侧弯、肘外翻、膝内翻、高弓腭、马德隆腕关节畸形、短掌骨、短趾骨、小颌畸形和感音神经性耳聋等[5-6]。与LWD相关的四肢比例失调、马德隆畸形等症状在幼儿阶段往往观察不到,具体的发病时间存在个体差异性,通常在青春期开始出现症状并逐渐加重[7-8]。芳香基硫酸酯酶E基因(ARSE)在Y染色体上无拷贝,并受到X染色体失活过程的影响。ARSE缺乏与X连锁隐性遗传斑点性软骨发育不全(CDPX)相关,临床表现为扁脸、鼻梁凹陷、鼻尖塌陷、身材矮小、肢体不对称性缩短和远端指(趾)发育不良,影像学可见软骨散在钙化[9]。胎儿的哥哥作为先证者目前出现了生长发育迟缓的情况,可能与SHOX、ARSE基因的缺失有关,比较遗憾的是先证者未行相关影像学检查,关于SHOX、ARSE基因缺失的其他骨骼异常在先证者上暂时未发现,但是不排除在以后的生长发育过程中出现。NLGN4X基因编码神经连接蛋白4,定位于X染色体短臂Xp22.33位置,该基因缺失与自闭症、智力低下、多动症、活动和注意力失调有关[10]。Xp/Yq易位的女性患者因为另一条X染色体上存在正常的NLGN4X基因一般不会出现智力低下。本研究中胎儿母亲智力正常,但是胎儿哥哥出现了智力发育迟缓、语言表达能力差、多动症、注意力维持短等相关症状。类固醇硫酸酯酶(STS)基因缺失可引起X-连锁鱼鳞病(XLI),是一种以皮肤广泛对称附着多角性干燥鳞屑为特征的X连锁隐性遗传病。患者可表现为全身或局部皮肤干燥、粗糙、黄褐色或污黑色鳞屑,腹部较背部严重[11]。另有研究表明,孕有XLI胎儿的孕妇,血清中仅存在低水平的uE3,血清学筛查对X-连锁鱼鳞病胎儿可能有一定提示作用[12]。本研究中孕妇前来就诊的原因之一是血清学筛查提示uE3中位数低,同时先证者又出现了皮肤鱼鳞病症状,与上述研究[12]相符。KAL1基因,又称ANOS1基因,定位于X染色体短臂Xp22.33位置,缺失可致Kallmann综合征(KS),表现为伴有嗅觉缺失或减退的性腺功能减退合并失眠、泌尿生殖系统异常(30%~40%为肾发育不良)和联带运动,但不导致其他发育缺陷如中线颅骨异常(唇裂、腭裂和不完全融合)[13]。本研究中先证者双眼屈光异常、睡眠障碍、小阴茎、隐睾,与文献报道[13]相符,患者没有进一步行泌尿生殖系统方面的超声检查,不确定是否存在肾脏方面相关问题。
通过文献检索,发现9例相似Xp/Yq不平衡易位男性患者的报道。每例患者根据XY断裂位点和重复、缺失片段大小不同,所表现出的临床症状也有所不同,见表1。病例1孕42周出生,生长发育迟缓伴有心内膜垫缺失、面部畸形、双手掌横向折痕、隐睾、皮肤鱼鳞病等多发异常,3个半月时死于心力衰竭[14]。病例2足月剖宫产,胎儿期超声检查发现肾脏异常,出生后诊断为多囊肾,轻度生长发育迟缓伴轻度智力障碍、面部畸形、耳朵畸形、手掌横向折痕、隐睾等多发畸形,未观察到明显的皮肤病变[15]。病例3、4患者年龄较大,具有正常的表型和身材,因不育症就诊,后续检查发现无精子症,两患者均存在心理障碍,未见其他异常[16]。病例5孕42周出生,生长发育迟缓、面部畸形、头发稀疏、中掌骨短、腹部皮肤鱼鳞病等[17]。病例6足月顺产,生长发育明显迟缓伴脑畸形、语言发育障碍、精神运动障碍、隐睾、皮肤鱼鳞病等[18]。病例7、8为两兄弟,哥哥就诊时24岁,身高163 cm,鼻梁塌陷,骨骼马德隆畸形,手腕和膝关节运动受损,婴儿期发现有鱼鳞病持续到现在,8岁时诊断癫痫发作,自儿童时期开始睡眠紊乱(间歇性的白天嗜睡和夜间失眠),活动与注意力失调,语言发育迟缓,学习困难;弟弟就诊时23岁,身高157 cm,面部畸形、塌鼻,骨骼马德隆畸形,尺骨远端突出,前臂旋后受限,皮肤鱼鳞病,2岁时因斜视接受眼部手术,11岁时诊断癫痫发作,和哥哥一样自童年开始的睡眠紊乱,活动与注意力障碍,学习困难,兄弟俩通过特殊教育完成了高中学业,目前都没有稳定的工作[19]。病例9患者智力发育障碍、脑瘫、隐睾,其他暂未见异常[20]。
总的来说,由Xp和Yq的同源序列之间的易位产生的衍生X染色体在人类中很少发生,目前仅报道了少数病例,大部分具有家族性。对于携带相似Xp/Yq不平衡易位的患者,由于Xp22.3区域的基因大多数为X连锁隐性遗传模式,异常主要发生在男性患者。男性患者因为Xp22.3区域的缺失常出现比较明显的连续基因综合征的复杂表型或死胎,具体临床表现随染色体断裂位点、重排片段大小、基因的缺失不同而有所不同。主要表现有发育迟缓、身材矮小、智力障碍、精神运动障碍、眼白化病、全身或局部皮肤鱼鳞病、隐睾、骨骼异常以及面部畸形如塌鼻、鼻短等。文献报道中的病例7、8成长过程中陆续出现不同症状表明Xp/Yq不平衡易位患者相关临床症状可能会随着生长发育慢慢凸显,对于该类染色体病患儿,即使幼儿期症状轻微,在其后续的成长过程还需密切关注,及时干预,改善生活质量。
传统的染色体核型G显带分析能对大部分的染色体数目和结构异常作出诊断,但对衍生染色体的性质、来源常难以识别,容易造成误诊。本研究中,如果单纯通过G显带技术,肉眼只能观察到X染色体短臂末端出现一浓染固缩状来源不明的片段,对于X染色体的具体异常不能确定,由于Yq大部分为异染色质,在G显带中多深染着色,而Xq也有深染条带,使得该衍生X染色体的形状类似于i(X)(q10),在分辨率水平较差的情况下容易误诊为i(X)(q10),胎儿哥哥在外院的染色体G显带结果就出现了类似的误诊。而染色体微阵列检测技术能够在全基因组水平进行扫描,可检测染色体不平衡的拷贝数变异,尤其对于检测染色体微小缺失、重复等不平衡性重排具有突出优势,弥补了染色体G显带分辨率的不足,从基因水平对异常染色体精确定位[21],同时染色体G显带可检出平衡易位及嵌合体等,两者结合可进一步减少漏诊误诊[22]。最终本研究通过染色体G显带/C显带检查结合染色体微阵列检测技术,提示X/Y染色体间的不平衡易位,并得出明确的断裂位点以及重复和缺失片段的大小。染色体断裂位点不同,重排片段大小不同,基因重复及缺失不同,胎儿性别不同等对胚胎的发育以及出生后生长情况的影响也不同。经优生遗传咨询,鉴于男性患者可能会有比较严重的临床表现以及哥哥作为先证者已经出现的种种临床表现,本研究中孕妇选择终止妊娠。
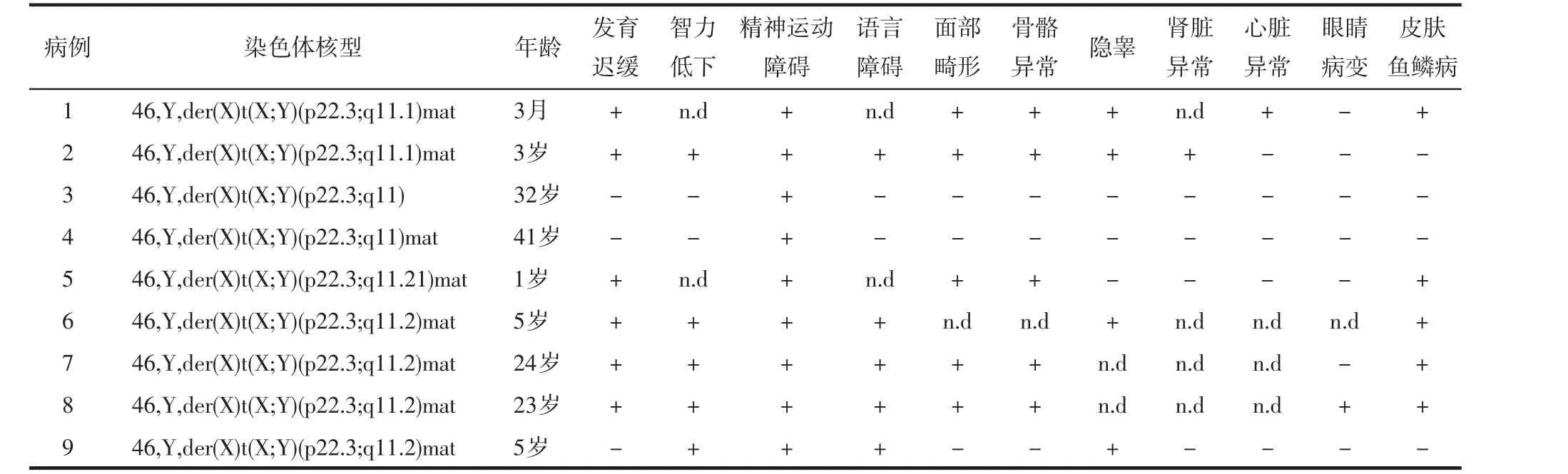
表1 9例Xp/Yq不平衡易位男性患者的临床资料
综上所述,目前的产前诊断技术平台能有效防止染色体病患儿的出生,染色体核型分析仍是国内染色体疾病产前诊断的金标准,而染色体微阵列检测技术的应用可以提示明确的断裂位点以及重复和缺失片段的大小,进一步提高了染色体疾病的诊断水平,更好地预测胎儿发生畸形的风险及准确地判断预后。建议同时联合染色体核型和染色体微阵列检测,从基因水平为临床遗传咨询提供指导,减少出生缺陷的发生。
