炒王不留行爆花与僵子差异性研究
2020-06-05林伟雄邓李红张志鹏罗宇琴
林伟雄,魏 梅,邓李红,张志鹏,罗宇琴,鲁 云
(广东一方制药有限公司/广东省中药配方颗粒企业重点实验室,广东 佛山 528244)
王不留行是石竹科植物麦蓝菜Vaccariasegetalis(Neck.)Garcke的干燥成熟种子,为我国传统常用中药,始载于《神农本草经》,列为上品[1],具有活血通经、下乳消肿、利尿通淋的功效;主要用于治疗经闭、痛经、乳汁不下、淋症涩痛等症[2]。研究显示,王不留行主要化学成分为黄酮苷、生物碱、环肽类、三萜皂苷类、挥发油等[3-4],其中王不留行黄酮苷和刺桐碱是其主要有效成分,具有促进乳汁分泌、保护血管内皮细胞、抗炎、增强免疫和抗肝损伤等药理作用[5-7]。
王不留行由于种子质地坚硬,有效成分不易煎出,炒法是王不留行自明代开始一直沿用至今的炮制工艺,炒制后的炒王不留行质地松泡利于有效成分煎出且走散力强,长于活血通经、下乳、通淋[8]。近年来,多数学者对王不留行炮制前后的区别进行了大量的研究,认为炮制后能提高王不留行有效成分水溶出率[5],但未见对王不留行炒制过程中未爆开白花的僵化现象进行研究,受限于当前生产炒药技术与设备水平,大生产机炒僵化现象比较常见,僵子入药对炒王不留行主要指标成分含量和整体功效是否存在影响是个值得探讨的话题,本实验对炒王不留行爆花和僵子饮片水溶性浸出物和主要指标成分含量进行测定和比较,同时结合现代先进UPLC技术,建立王不留行炒制前后UPLC特征图谱,快速全面比较炒王不留行爆花和僵子饮片的差异性,建立王不留行及其炮制品饮片的更高质量控制方法,更好地指导生产和保证临床疗效。
1 仪器与试药
1.1 仪器
Waters高效液相色谱仪(沃特世公司,Waters e2695);Waters超高效液相色谱仪(沃特世公司,Waters H-class);TUV检测器(沃特世公司);Empower工作站;ME204E型万分之一天平(梅特勒-托利多公司);XP26型百万分之一天平(梅特勒-托利多公司);Milli-Q Direct 8/16 system型超纯水机(默克公司);KQ5500DE型超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药
王不留行黄酮苷对照品(中国食品药品检定研究院,纯度>99.7%,批号:111853-201704),刺桐碱对照品(四川省维克奇生物科技有限公司,纯度>98.0%,批号:wkq18012302)。液相用甲醇、乙腈(默克股份有限公司)为色谱级,水为超纯水(默克股份有限公司,Mili-Q Direct),其余试剂为分析纯。
王不留行药材信息详见表1,经广东一方制药有限公司魏梅主任药师鉴定为正品,符合《中国药典》2015版一部“王不留行”项下相关规定。选取其中全国主要产区河北省的3批王不留行药材Y18、Y19、Y20,作为炒王不留行爆花和僵子研究用。
2 方法与结果
2.1 样品炮制
取净王不留行置炒药机内,按《中国药典》2015版清炒法(通则0213)炒至大多数爆开白花,呈类球形爆花状,表面白色,质松脆,取出,晾凉,筛去灰屑,得“炒王不留行饮片”。根据是否爆开白花将其分为“爆花饮片”和“僵子饮片”。

表1 王不留行药材来源
2.2 浸出物测定
分别称取样品约2 g,以水为溶剂,照《中国药典》2015版浸出物测定法(通则2201)项下的热浸法测定,测定结果见表6。
2.3 王不留行黄酮苷含量测定
2.3.1 色谱条件 Thermo Acclaim C18色谱柱(4.6 mm×250 mm,5 μm);流动相甲醇(A)-0.3%磷酸(B),梯度洗脱(0~10 min,35% A;10~20 min,35%~40% A;20~35 min,40%~50% A),检测波长280 nm,进样量10 μL,流速0.7 mL·min-1,柱温30 ℃。
2.3.2 对照品溶液制备 取王不留行黄酮苷对照品适量,精密称定,置10 mL量瓶中,加70%甲醇定容至刻度,摇匀,即得每1 mL含王不留行黄酮苷0.772 8 mg的储备液,精密量取储备液2 mL于10 mL量瓶中,加70%甲醇制成每1 mL含154.556 μg的溶液,摇匀,即得。
2.3.3 供试品溶液制备 取本品粉末(过三号筛)约1.2 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50 mL,称定重量,超声处理(功率250 W,频率33 kHz)30 min,放冷,再称定重量,用70%甲醇补足减失重量,摇匀,滤过,取续滤液,作为供试品溶液。
2.3.4 方法学考察 (1)专属性试验。精密吸取王不留行黄酮苷对照品溶液、空白溶剂和“2.3.3”项下供试品溶液,按照“2.3.1”项下色谱条件测定,结果如图1所示。
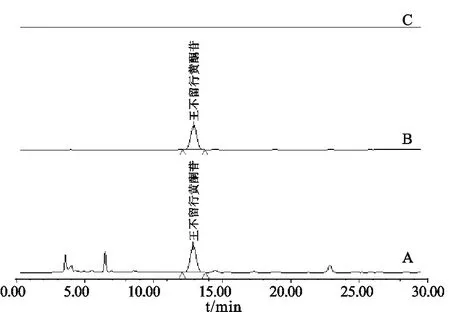
注:A.供试品溶液;B.王不留行黄酮苷对照品;C.空白溶剂
(2)线性关系及范围。精密量取上述王不留行黄酮苷对照品储备液10.0、6.0、2.0、1.0、0.5、0.1 mL,分别置10 mL量瓶中,加70%甲醇定容至刻度,摇匀,配制成7.728、38.639、77.278、154.556、463.667、772.778 μg/mL的对照品溶液,分别按照“2.3.1”项下色谱条件进样测定,以峰面积为纵坐标(Y),对照品浓度为横坐标(X),得王不留行黄酮苷回归方程为Y=8 859X+5 253.8(R2=0.999 9),表明在浓度为7.728 ~772.778 μg·min-1的范围内王不留行黄酮苷浓度与峰面积线性关系良好。

表2 王不留行黄酮苷标准曲线考察
(3)精密度试验。精密吸取王不留行黄酮苷对照品溶液,按照“2.3.1”项下色谱条件连续进样6次。计算王不留行黄酮苷峰面积的RSD值为0.45%,表明仪器精密度良好。
(4)稳定性试验。精密吸取同一供试品溶液,分别于配制后0、2、4、6、8、12 h按照“2.3.1”项下色谱条件进样测定,计算王不留行黄酮苷峰面积的RSD值为0.52%,表明供试品溶液中王不留行黄酮苷在12 h内稳定。
(5)重复性试验。精密称取同一批样品6份,按“2.3.3”项下确定的供试品溶液制备方法,分别制备王不留行黄酮苷含量测定供试品溶液6份。按“2.3.1”项下色谱条件进样测定,结果王不留行黄酮苷含量的RSD值为0.30%,表明该方法重复性良好。
(6)加样回收率试验。取已测定含量的王不留行粉末约0.6 g,精密称定,分别精密加入一定量的王不留行黄酮苷对照品,按“2.3.3”项下供试品溶液制备方法,制备供试品溶液9份。按“2.3.1”项下色谱条件测定,计算王不留行黄酮苷加样回收率,结果见表3,平均回收率为99.42%,RSD值为1.35%,表明回收率良好。

表3 王不留行中王不留行黄酮苷加样回收试验结果 (n=9)
2.3.5 样品测定 将3批实验用生品、爆花和僵子饮片分别按照“2.3.3”项下供试品溶液制备方法制备相应的供试品溶液,并按“2.3.1”项下色谱条件进样测定,并计算炮制后爆花和僵子饮片王不留行黄酮苷含量损失情况。结果见表6。
2.4 刺桐碱含量测定
2.4.1 色谱条件 YMC C18色谱柱(2.1 mm×100 mm,1.9 μm);流动相乙腈(A)-水(B),梯度洗脱(0~5.5 min,10%~15% A;5.5~11 min,15%~30% A;11~16.5 min,30%~70% A;16.5~16.51 min,70%~10% A;16.51~23 min,10% A),检测波长219 nm,进样量1 μL,流速0.35 mL·min-1,柱温35 ℃。
2.4.2 对照品溶液制备 取刺桐碱对照品适量,精密称定,置25 mL量瓶中,加甲醇定容至刻度,摇匀,即得每1 mL含刺桐碱0.160 9 mg的储备液,精密量取储备液2.5 mL于10 mL量瓶中,加甲醇制成每1 mL含40.229 μg的溶液,摇匀,即得。
2.4.3 供试品溶液制备 取本品粉末(过三号筛)约1.0 g,精密称定,置具塞锥形瓶中,精密加入75%乙醇25 mL,称定重量,超声处理(功率250 W,频率33 kHz)30 min,放冷,再称定重量,用75%乙醇补足减失重量,摇匀,滤过,取续滤液,作为供试品溶液。
2.4.4 方法学考察 (1)专属性考察。精密吸取刺桐碱对照品溶液、空白溶剂和“2.4.3”项下供试品溶液,按照“2.4.1”项下色谱条件测定,结果如图2所示。
(2)线性关系考察。精密量取上述刺桐碱对照品储备液10.0、5.0、2.5、2.0、1.0、0.5 mL,置10 mL量瓶中,加甲醇定容至刻度,摇匀,配制成8.046、16.092、32.183、40.229、81.458、160.916 μg/mL的对照品溶液,分别按照“2.4.1”项下色谱条件依次进样测定;以峰面积为纵坐标(Y),对照品浓度为横坐标(X),得刺桐碱的回归方程为Y=19 677X-21 603(R2=0.999 7),表明在浓度为8.046~160.916 μg·min-1范围内刺桐碱浓度与峰面积线性关系良好。

表4 刺桐碱标准曲线考察

注:A.供试品溶液;B.刺桐碱对照品;C.空白溶剂
(3)精密度试验。精密吸取刺桐碱对照品溶液,按照“2.4.1”项下色谱条件连续进样6次。计算刺桐碱峰面积的RSD值为0.62%,表明仪器精密度良好。
(4)稳定性试验。精密吸取同一供试品溶液,分别于配制后0、2、4、6、8、12 h按照“2.4.1”项下色谱条件进样测定,计算刺桐碱峰面积的RSD值为1.11%,表明供试品溶液中刺桐碱在12 h内稳定。
(5)重复性试验。精密称取同一批样品6份,按“2.4.3”项下确定的供试品溶液制备方法,分别制备刺桐碱含量测定供试品溶液6份。按“2.4.1”项下色谱条件进样测定,结果刺桐碱的含量RSD值为1.06%,表明该方法重复性良好。
(6)加样回收率试验。取已测定含量的王不留行粉末约0.5 g,精密称定,分别精密加入一定量的刺桐碱对照品,按“2.4.3”项下供试品溶液制备方法,制备供试品溶液9份。按“2.4.1”项下色谱条件测定,计算刺桐碱加样回收率,结果见表5,平均回收率为96.68%,RSD值为2.40%,表明回收率良好。
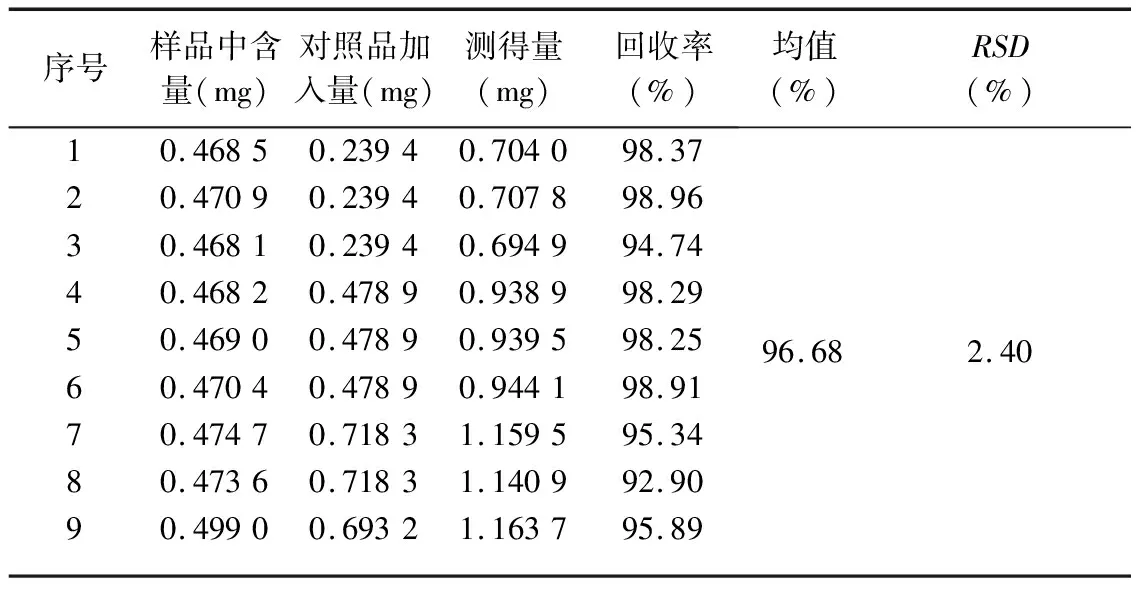
表5 王不留行中刺桐碱加样回收试验结果 (n=9)
2.4.5 样品测定 将3批实验用生品、爆花和僵子饮片分别按照“2.4.3”项下供试品溶液制备方法制备相应的供试品溶液,并按“2.4.1”项下色谱条件进样测定,并计算炮制后爆花和僵子饮片刺桐碱含量损失情况。结果见表6。
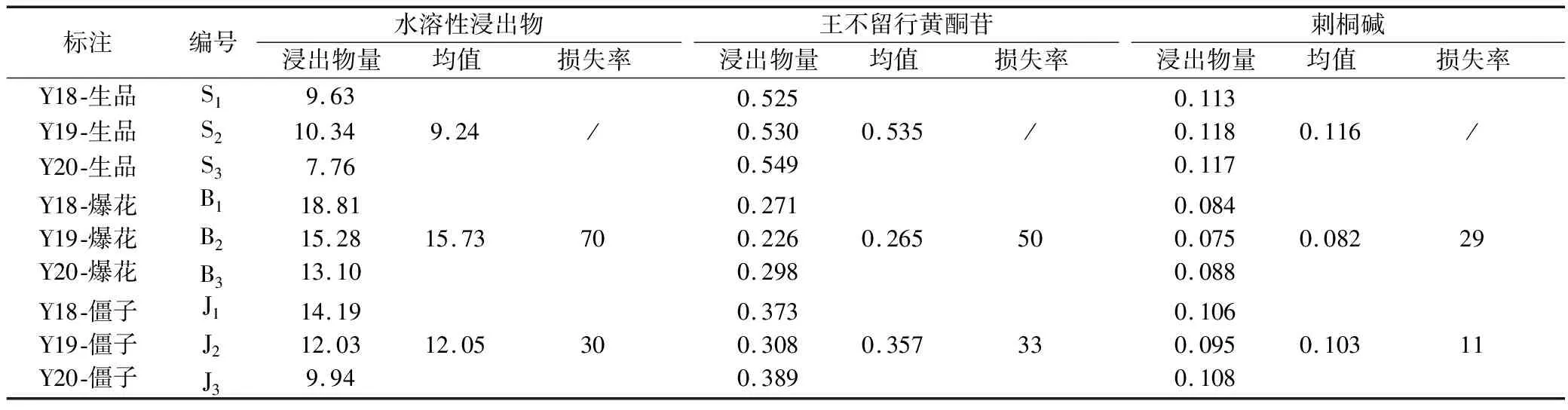
表6 王不留行炮制前后变化情况 (%)
注:提高溶出率%=(饮片浸出物均值-生品浸出物均值)/生品浸出物均值×100%;损失率%=(1-饮片含量均值/生品含量均值)×100%。
浸出物结果显示,炒制后饮片均能明显增大生品的水溶性浸出物,爆花饮片能提高生品约70%的水溶出率,这也验证了前人研究结论[5],而僵子饮片只能提高生品约30%水溶出率,炒制僵化现象会降低合格炒王不留行爆花饮片的水溶出率。王不留行生品、炒制后爆花和僵子饮片的王不留行黄酮苷含量均值分别为0.535%、0.265%、0.357%,刺桐碱含量均值分别为0.116%、0.082%、0.103%。炒制后主要指标成分含量均出现明显下降,炒制过程中含量不断损失,爆花饮片中王不留行黄酮苷和刺桐碱含量分别损失50%和29%,而僵子饮片中相应损失33%和11%,炒制后爆花饮片主要指标成分损失率明显大于僵子饮片。
2.5 特征图谱测定
2.5.1 特征图谱建立 取20个批次的王不留行药材,按“2.4.3”项下方法制备供试品溶液,按“2.4.1”条件测定并记录色谱图,分别导入《中药色谱指纹图谱相似度评价软件》(2012A)进行比较分析,如图3所示,共确定了5个共有特征峰,其中,与对照品对照指认了峰1为刺桐碱,峰2为王不留行黄酮苷。以2号峰王不留行黄酮苷作为参照峰(S峰),计算各共有特征峰相对保留时间和相对峰面积,按照特征图谱研究技术的要求,生成王不留行共有对照特征图谱并建立王不留行UPLC特征图谱,如图4所示。
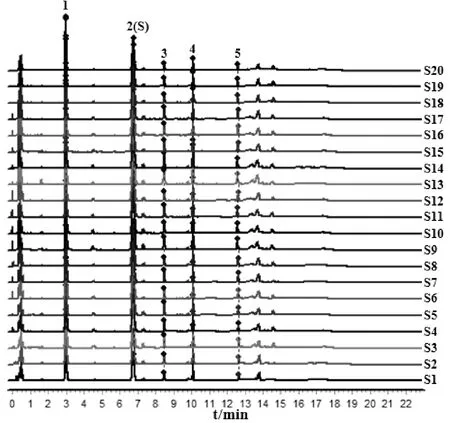
注:1.刺桐碱;2(S).王不留行黄酮苷
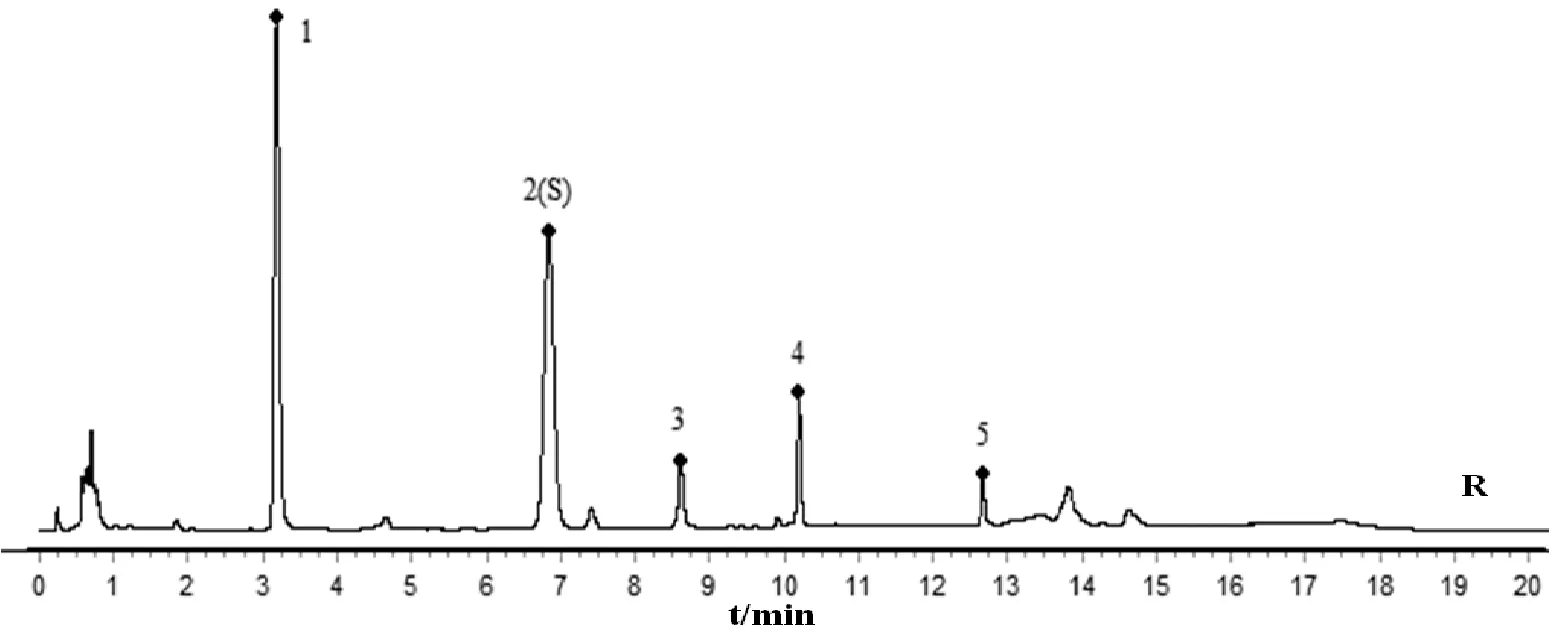
注:1.刺桐碱;2(S).王不留行黄酮苷
2.5.2 特征图谱方法学考察 (1)精密度试验。同“2.4.3”项下测定,记录色谱图,以2号峰王不留行黄酮苷的色谱峰为参照峰,对各图谱的特征峰的相对保留时间和相对峰面积进行计算,结果各主要色谱峰的相对保留时间和相对峰面积无明显变化,其RSD值分别为0.00%~0.10%和0.00%~0.08%,表明仪器精密度良好,符合特征图谱研究技术的要求。
(2)稳定性试验。同“2.4.3”项下(4)测定,记录色谱图,以2号峰王不留行黄酮苷的色谱峰为参照峰,对各图谱的特征峰的相对保留时间和相对峰面积进行计算,结果各主要色谱峰的相对保留时间和相对峰面积无明显变化,其RSD值分别为0.00%~0.33%和0.00%~0.52%,表明供试品溶液在12 h内稳定性良好,符合特征图谱研究技术的要求。
(3)重复性试验。同“2.4.3”项下(5)测定,记录色谱图,以2号峰王不留行黄酮苷的色谱峰为参照峰,对各图谱的特征峰的相对保留时间和相对峰面积进行计算,结果各主要色谱峰的相对保留时间和相对峰面积无明显变化,其RSD值分别为0.00%~0.24%和0.00%~0.14%,表明该方法重复性良好,符合特征图谱研究技术的要求。
2.5.3 特征图谱检测与分析 将3批实验用生品、爆花和僵子饮片分别按照“2.4.3”项下方法制备供试品溶液,按“2.4.1”条件测定并记录色谱图,统计各共有峰特征峰保留时间和峰面积,见表7-表8,分别导入《中药色谱指纹图谱相似度评价软件》(2012A)进行特征图谱比较分析。如图5所示,以峰2王不留行黄酮苷作为参照峰,计算各共有峰特征峰相对保留时间和相对峰面积,见表9-表10;并计算各样品与20批王不留行药材生成的共有对照特征图谱间相似度,见表11;同时,将王不留行生品、爆花和僵子饮片分别生成共有对照指纹图谱进行直观比较,如图6所示。

表7 共有特征峰保留时间 (min)
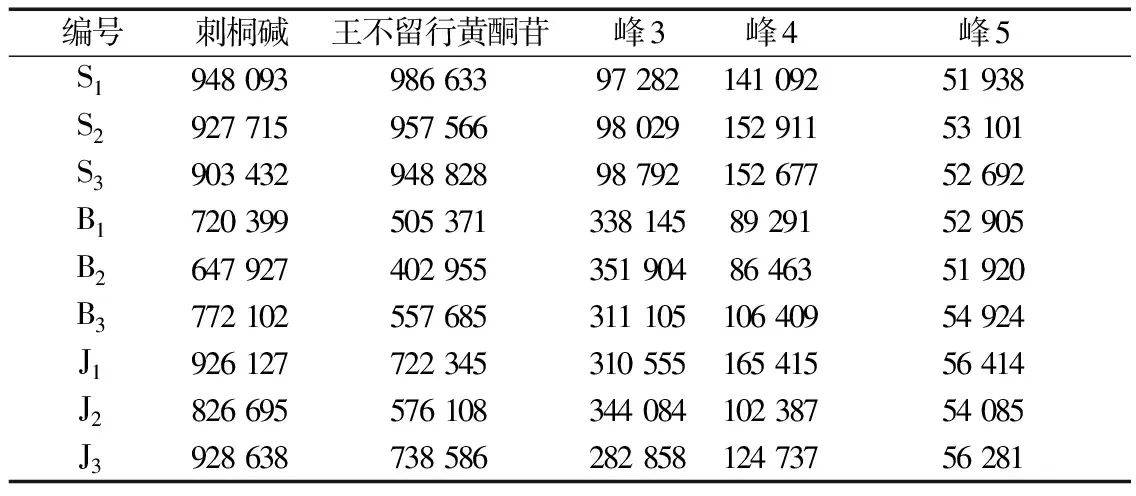
表8 共有特征峰峰面积
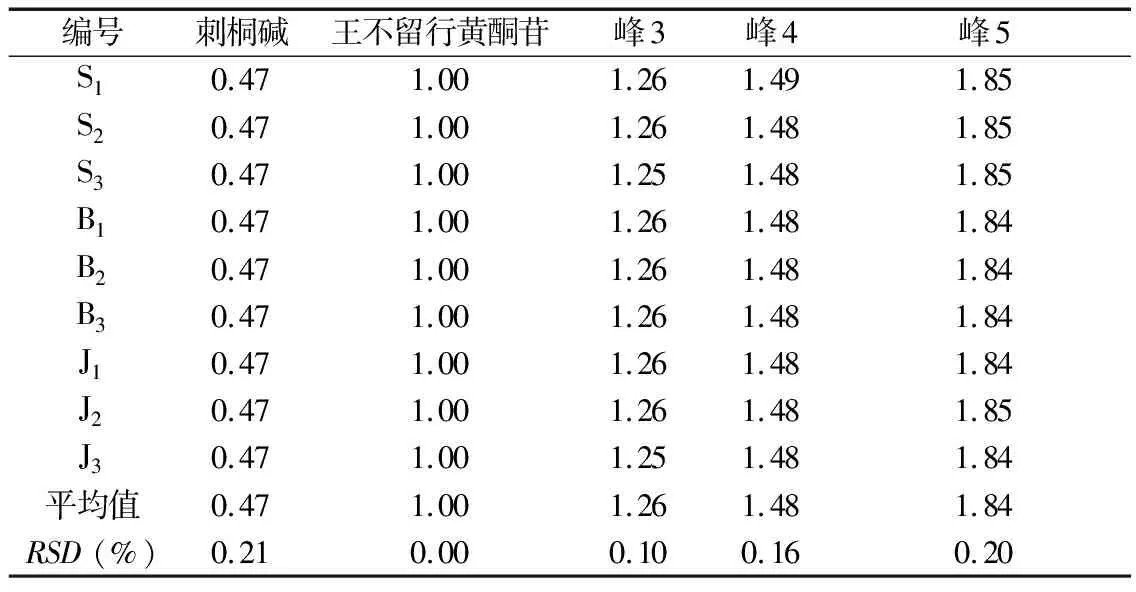
表9 共有特征峰相对保留时间 (min)

表10 共有特征峰相对峰面积
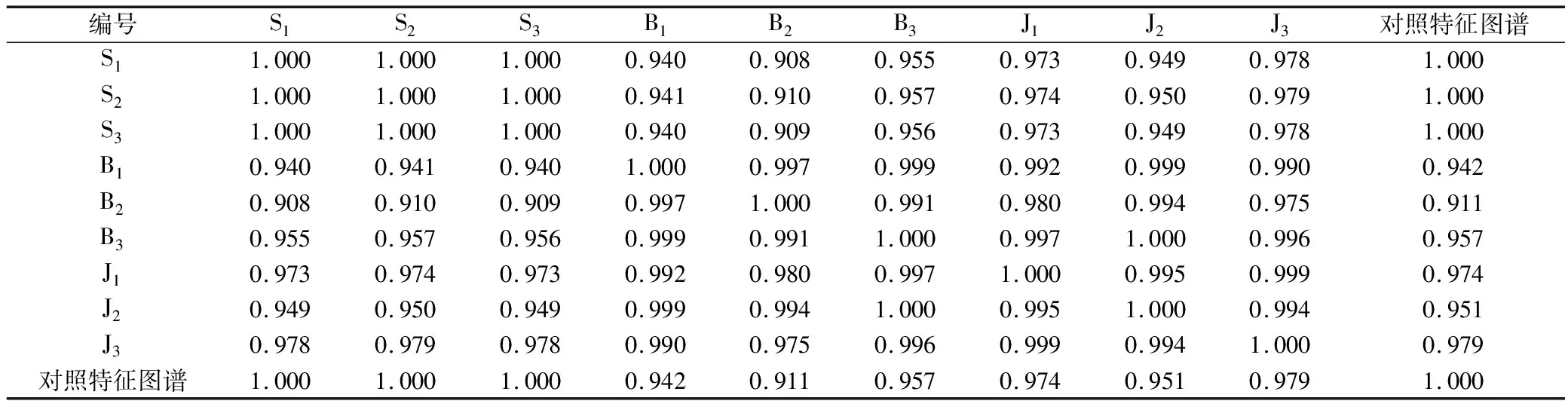
表11 相似度计算结果
实验结果显示,王不留行生品、炒制后爆花和僵子饮片各共有峰特征峰保留时间和相对保留时间较稳定,而各共有峰特征峰的峰面积和相对峰面积发生了明显变化。炒制后,峰1、2、4峰面积均明显降低,且爆花饮片下降比僵子饮片明显,而炒制后峰3峰面积明显变大,但爆花和僵子饮片间差异性不大,均增大约2倍,峰5则基本不变,较为稳定。
各样品与20批王不留行药材生成的共有对照特征图谱间相似度均在0.90以上,相似度较高,表明炒制前后主要化学成分和色谱峰的数量基本一致,如图6直观可见,但爆花饮片与生成的共有对照特征图谱间的相似度均低于僵子,表明炮制后爆花饮片各色谱峰变化程度大于僵子。
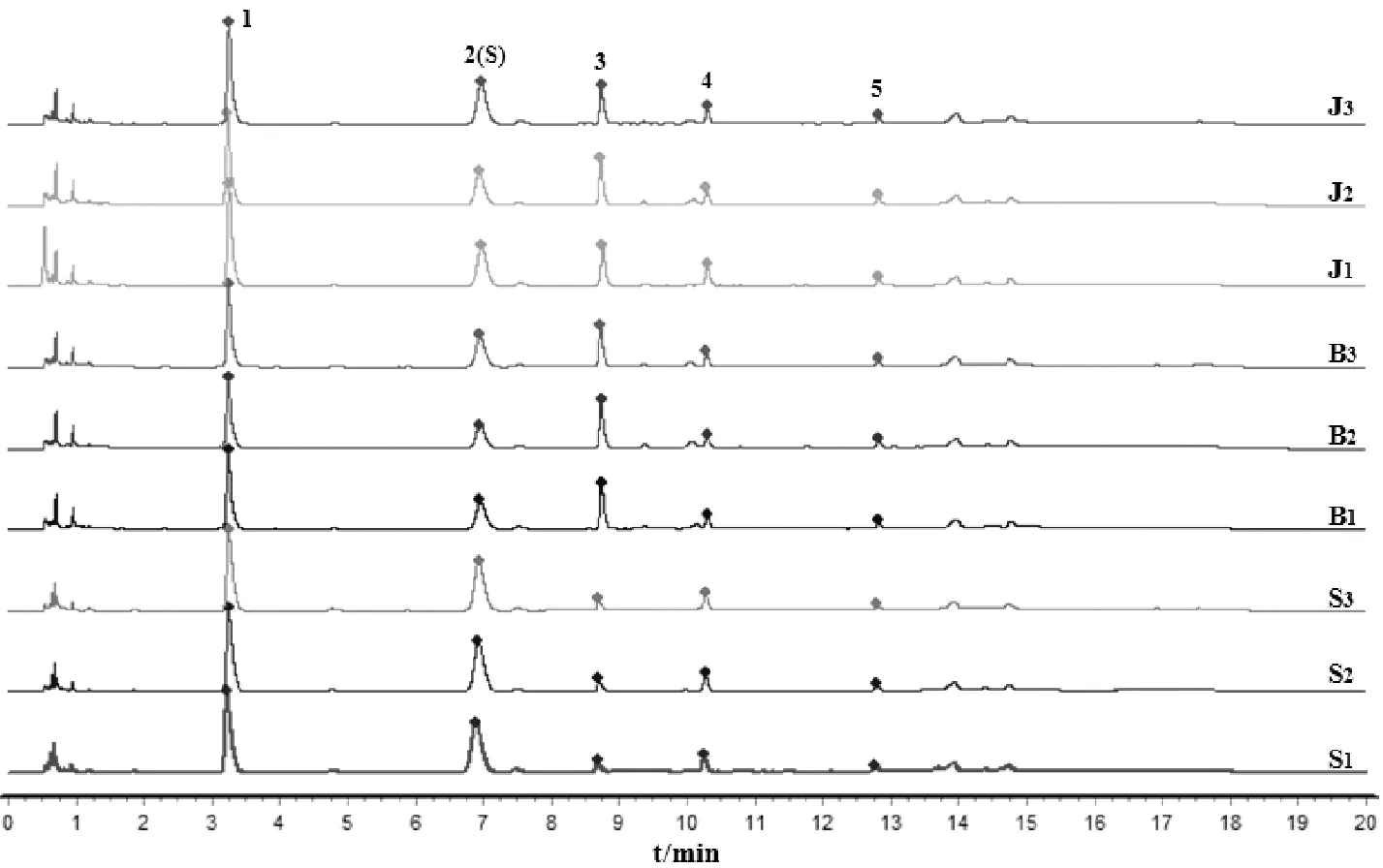
注:S1-S3为生品;B1-B3为爆花;J1-J3为僵子
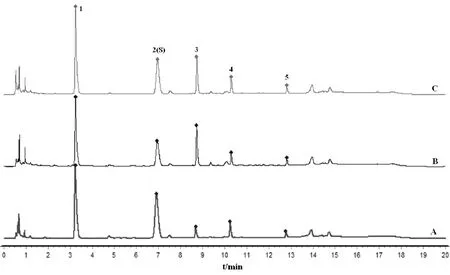
注:A为生品;B为爆花;C为僵子
3 讨论
实验结果表明,炒制后饮片均能明显增大生品的水溶性浸出物,但爆花饮片对生品的水溶出率提高能力约为僵子饮片的2倍;炒制后主要指标成分含量均出现明显下降,炒制过程中含量不断损失,爆花饮片主要指标成分损失量明显大于僵子饮片,可能是僵化现象减少了指标成分的破坏损失。《中国药典》2015年版王不留行药材含量测定项规定王不留行黄酮苷不得少于0.40%,炒王不留行饮片含量测定项规定王不留行黄酮苷不得少于0.15%,也说明该成分炮制后是会明显降低,但未对炮制品含量上限作出进一步规定。受限于当前生产炒药技术和设备水平,机炒王不留行的爆花率偏低一直是中药炮制的重难点之一,僵化现象较为常见,炒王不留行饮片中混有较多僵子,其王不留行黄酮苷的含量会明显高于完全爆开白花饮片的含量,出现炮制品含量符合药典要求,但实际饮片性状不符合药典要求的情况。特征图谱结果显示,炒制后爆花和僵子饮片峰1、2、4峰面积均明显降低,且爆花饮片降低幅度均大于僵子饮片,爆花饮片与生品相似度较小,说明两者间差异性较大。总体来看,炮制合格爆花饮片从水溶性浸出物、主要指标成分含量、特征图谱总体情况等与生品间的差异性较大,这也是王不留行生品炮制可以明显提高其活血通经、下乳、通淋的作用其意义所在,而僵子与生品差异性较小,主要指标成分含量虽然合格,但会影响炒王不留行合格饮片的水溶出率,降低炒王不留行水提过程有效成分的煎出,同时也影响特征图谱中部分峰的变化,僵子饮片可能难以发挥炒制合格的爆花饮片长于王不留行生品的功效,有待进一步研究。
炮制作为中药的精华和特色,对保证中药临床疗效具有十分重要的作用。中医医师在临证处方时,常根据不同病情选用不同炮制品,有目的地运用以提高临床疗效。因此,炒王不留行不同爆花程度的饮片选择和质量控制对中医临床的安全性和有效性至关重要。本实验建立了王不留行炮制前后UPLC特征图谱,分离效果好,重复性强,保留时间相对稳定,各共有特征峰含量变化明显,且可同时测定刺桐碱含量,能简单快速全面地控制王不留行炮制前后特征信息,并结合《中国药典》2015版“王不留行”项下王不留行黄酮苷含量测定方法,对王不留行炮制前后主要指标成分刺桐碱和王不留行黄酮苷进行双指标含量控制,更加全面系统地评价王不留行炮制前后及不同炒制程度爆花与僵子饮片的差异性,更好地保证饮片质量和临床疗效。
