1例复合杂合突变导致遗传性凝血因子Ⅺ缺陷症家系的基因分析
2020-06-05方伟伟郭跃丽奚经巧
方伟伟,郭跃丽,奚经巧
(温州市中医院,浙江 温州 325015)
凝血因子Ⅺ (FⅪ),又称为 “血浆凝血活酶前质”,主要在肝细胞和巨核细胞中产生合成。可通过凝血酶、活化的凝血因子Ⅻ或自身裂解所激活,生成活化的FⅪ(FⅪa),进而激活凝血因子Ⅸ,从而活化凝血级联反应。遗传性FⅪ缺陷症为常染色体隐性遗传,男女均可发病。患者临床出血表现轻重不一,主要表现为创伤或手术后出血增多,鲜有明显的自发性出血,且FⅪ活性(FⅪ:C)水平高低与出血程度并不相关[1]。目前认为该疾病的发生与FⅪ的基因突变有关[2]。本研究通过对一个复合杂合性(F11)基因突变导致的遗传性FⅪ缺陷症家系进行实验室表型检测与基因突变定位分析,初步探讨其分子致病机制。
1 病例介绍
先证者,男,28岁。2018年6月因腮腺良性肿瘤收住入院,术前凝血常规检查发现活化部分凝血活酶时间(APTT)为84.2秒,进一步检查发现FⅪ:C 为 3%,FⅪ抗原(FⅪ:Ag)为 8.6%。其他相关凝血指标均无明显异常,肝、肾功能正常,初步诊断为“FⅪ缺陷症”。征得先证者及其家系成员知情同意后调查先证者及其家系3代6人,均无自发性出血症状,先证者父母非近亲结婚,家系遗传图谱详见图1。正常对照:选择100例体检健康者,年龄22-56岁,无肝、肾功能异常,且无其他基础性疾病,其中男53例,女47例,用于排除基因多态性。
2 方法
2.1 样品采集与处理 采集受检者外周静脉血2.7mL,采用0.109mol/L枸橼酸钠1:9抗凝,3000r/min离心10分钟,留取上层乏血小板血浆用于凝血指标检测,下层血细胞用于提取基因组DNA。

图1 遗传性凝血因子Ⅺ缺陷症家系图
2.2 实验室表型检测 APTT、血浆凝血酶原时间(PT)、纤维蛋白原(FIB)、FⅪ:C、FⅧ活性(FⅧ:C)、FⅨ活性(FⅨ:C)、FⅫ活性(FⅫ:C)等凝血指标采用一期凝固法在法国STAGO STA-R全自动血凝仪上检测(配套试剂由法国STAGO公司提供);FⅪ:Ag采用酶联免疫吸附测定法检测。所有操作步骤均严格按照试剂说明书进行。
2.3 基因分析 DNA提取采用非离心柱型基因组DNA抽提试剂盒抽提先证者及家系成员的外周血基因组DNA。根据人类(F11)基因序列(GenBank AYl91837),应用primer 5.0软件共设计13对引物,以覆盖(F11)基因的15个外显子区域及其侧翼序列。引物序列及PCR反应条件参见文献[2]。PCR产物经1%琼脂糖凝胶电泳鉴定,阳性标本纯化后进行测序(上海桑尼生物科技有限公司)。将测序结果与Genbank 数据库中(F11)基因序列(NG_008051.1)比对,发现突变位点后反向测序予以证实。先扩增先证者(F11)基因各外显子、侧翼序列以及5′端、3′端非翻译区,发现突变位点后再扩增其他家系成员相应外显子片段。
2.4 氨基酸突变位点保守性分析 用ClustalX-2.1-win软件将突变氨基酸与其他同源物种:黑猩猩(Pan troglodytes)、猕猴(Macaca mulatta)、家犬(Canis lupus familiaris)、牛 (Bos Taurus)、小 家 鼠(Mus musculus)、 褐 鼠 (Rattus norvegicus)、 原 鸡(Gallus gallus)、热带爪蟾(Xenopus tropicalis)(同源物种氨基酸序列来源:http://www.ncbi.nlm.nih.gov/homologene)的氨基酸序列进行比对,分析突变氨基酸在物种进化过程中的保守性。
2.5 氨基酸突变前后生物信息学特性分析 用MutationTaster(http://www.mutationtaster.org)(蛋白质参考ID:P00747和蛋白质参考转录本ID:ENST00000308192)在线生物信息学软件,通过评分结果预测基因突变对蛋白质功能的影响,分数越接近1表明预测结果越可靠。
2.6 氨基酸突变前后蛋白质局部空间构,型变化分析 根据已知的FⅪ蛋白质结构模型构建突变后模型,针对突变位点用Swiss-PdbViewer软件分析(F11)基因突变前后由氨基酸变化引起的蛋白质空间结构的改变。
3 结果
3.1 实验室表型检测 先证者的APTT为84.2秒,明显延长,其FⅪ:C和FⅪ:Ag均明显下降,分别为3%和8.6%;家系成员中其父亲、母亲、祖父和外祖母的FⅪ:C和FⅪ:Ag均下降至参考值的一半左右,其母亲和外祖母的APTT稍延长于参考值,其他相关凝血指标均无明显异常。详见表1。
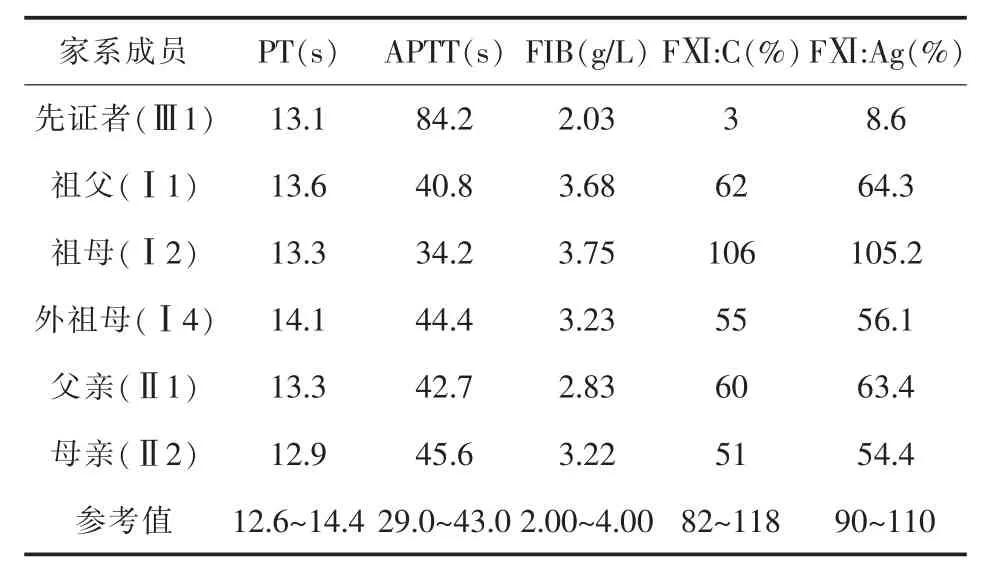
表1 先证者及家系成员实验室表型检测结果
3.2 基因分析 先证者(F11)的基因第7号外显子存在c.738G>A杂合无义突变导致p.Trp228stop,第12号外显子存在c.1325delT缺失突变导致p.L424CfsX8。其母亲和外祖母存在p.Trp228stop突变杂合子,其父亲和祖父存在p.L424CfsX8突变杂合子。详见图2、表2。
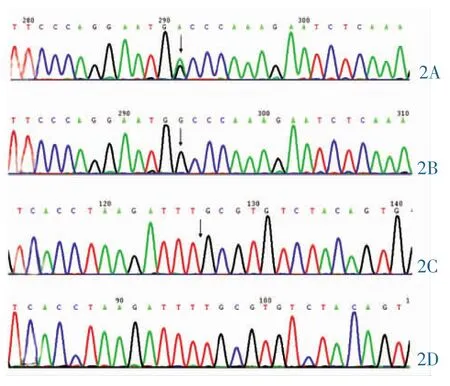
图2 (F11)基因第7和第12外显子测序结果。2A:c.738G>A 杂合无义突变;2B:c.738G 野生型;2C:c.1325delT缺失突变;2D:c.1325野生型。
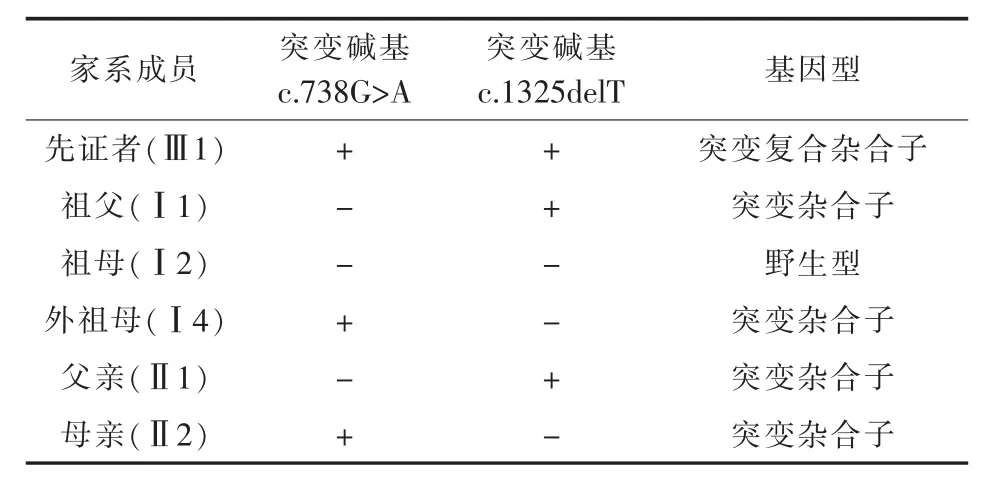
表2 先证者及家系成员基因分析结果
3.3 同源性分析 ClustalX-2.1-win软件保守性分析结果显示,Leu424在同源物种间呈高度保守。详见图3。
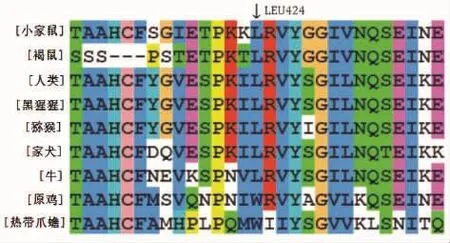
图3 Leu424同源性分析结果图
3.4 突变对蛋白影响力评估 Mutation Taster对p.Leu424Cys的评分结果为1.000分,预示此突变可引起相关疾病。
3.5 模型分析 Swiss-PdbViewer软件对FⅪ蛋白质进行模型分析发现,在野生型FⅪ蛋白质中非极性疏水性的Leu424与Pro421之间有一个氢键连接;当突变为极性亲水性的Cys424后,原有的氢键没有改变,但与Pro421的同一部位又多了一条氢键连接,导致蛋白空间结构发生改变。详见图4。
4 讨论
成熟的FⅪ分子是由两条多肽链通过二硫键连接形成的二聚体,FⅪ单链羧基端是胰蛋白酶样丝氨酸蛋白酶结构域(SP),氨基端由苹果状结构域(AP)组成。遗传性FⅪ缺陷症主要与(F11)基因突变有关,在其突变的数据(http://www.factorⅪ.com/)显示:突变分布于(F11)基因的各结构域,SP区的突变相对集中。FⅪ缺陷症在一般人群中的发生频率为1/10万~1/100万[3],而后续的研究数据显示:轻度FⅪ缺陷症的发生率比预期的要高[4]。FⅪ缺陷症的发病率与基因突变类型具有种族差异。该病在犹太人群中尤其高发,纯合突变患者比例可高达1/190。人类基因组数据库 (Human Gene Mutation Database,HGMD,http:/www.hgmd.cf.ac.uk/ac/a11.php)共收录(F11)基因突变 240余种,其中78.3%为错义突变和无义突变,9.8%为剪切位点突变,7.4%为小片段的缺失,剩余4.5%则为其它类型突变。遗传性FⅪ缺陷症分为2型,FⅪ:Ag和FⅪ:C同步下降,属于交叉反应物质(crossreation material,CRM)阴性型,若 FⅪ:C 降低而 FⅪ:Ag正常则为CRM阳性型。本文通过对一个遗传性FⅪ缺陷症家系进行实验室表型检测与基因突变分析,共发现了2个基因突变,分别为c.738G>A无义突变和c.1325delT缺失突变,均为CRM阴性型。
2003年武文漫等[3]首次报道位于AP3的c.738G>A 杂合无义突变(p.Trp228stop)。Shao等[5]发现该突变在57个中国FⅪ缺陷症家系最为常见,是中国人群FⅪ缺陷症的热点突变,且为中国人所特有。p.Trp228stop突变翻译而成的是截短型蛋白,高度不稳定并迅速被降解[6]。但也有研究认为,该无义突变产生的含有提前终止密码子的mRNA可被一种称之为 “无义介导衰变的mRNA监视系统”迅速降解,使得循环中FⅪ减少[7]。
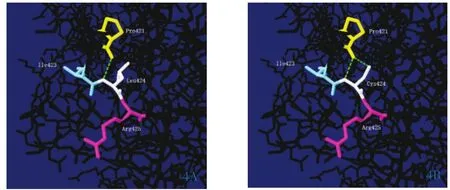
图4 FⅪ蛋白质模型图。4A:野生型;4B:突变型;绿色虚线为氢键。
Leu424位于第12号外显子SP区域,c.1325delT即1325号位置的T碱基缺失导致424后所有的氨基酸发生移码突变。氨基酸保守性分析显示,Leu424残基在同源物种间呈高度保守,提示它是FⅪ分子的一个重要功能性位点。MutationTaster生物信息学预测软件显示此突变为有害突变,可对蛋白质功能造成一定的影响,从而引起相应疾病。通过构建氨基酸突变前后结构模型可见424位Leu被Cys所替代并且在431位出现终止密码子,导致编码蛋白翻译提前终止而产生截短蛋白。该蛋白因缺失部分结构域,高度不稳定,功能域的缺失及不稳定性的增强影响了催化活性区[6],这可能是FⅪ:C及FⅪ:Ag下降的原因。
先证者基因分析显示,(F11)基因第7号外显子存在c.738G>A杂合无义突变导致p.Trp228stop,及第12号外显子c.1325delT缺失突变导致p.L424CfsX8。其母亲存在p.Trp228stop突变杂合子,其父亲存在p.L424CfsX8突变杂合子,结合家系图,先证者的p.Trp228stop杂合突变遗传自其母亲,p.L424CfsX8杂合突变遗传自其父亲。其父母为单一的杂合突变,FⅪ抗原与活性水平均降低至正常范围的一半左右,双重杂合突变导致先证者FⅪ抗原与活性水平极度降低,因此,该复合杂合突变与先证者FⅪ水平降低有关。有报道指出遗传性FⅪ缺陷症的单一纯合突变和双重杂合突变都可导致血浆FⅪ:C下降至15%以下,单一杂合突变的下降程度为15%~70%[8],与本文家系中有基因缺陷的家系成员FⅪ:C下降程度一致。本文100例正常对照者的基因均未发现p.L424CfsX8,排除了基因多态性的可能,同时查阅相关网站和文献,p.L424CfsX8突变尚未见报道。
综上所述,本文通过对一个遗传性FⅪ缺陷症家系进行实验室表型检测和基因突变分析,发现了p.Trp228stop杂合无义突变和p.L424CfsX8杂合缺失突变,后者为国内外首次报道。该家系人员FⅪ水平减低与这两种突变有关,但其确切的致病机制有待进一步研究。
